ADVANCED MATERIALS
This fits with a mechanism in which positively charged LNPs first bind to fibrinogen, inducing a conformational change in the protein that activates platelets, which in turn activates the rest of ..


Physicochemical Targeting of Lipid Nanoparticles to the Lungs Induces Clotting: Mechanisms and Solutions
Serena Omo-Lamai, Marco E. Zamora, Manthan N. Patel, Jichuan Wu, Jia Nong, Zhicheng Wang, Alina Peshkova, Aparajeeta Majumder, Jilian R. Melamed, Liam S. Chase … See all authors
First published: 23 February 2024
https://doi.org/10.1002/adma.202312026Digital Object Identifier (DOI)
Sections
Abstract
Lipid nanoparticles (LNPs) have become the dominant drug
delivery technology in industry, holding the promise to deliver RNA to
up or down-regulate any protein of interest. LNPs have mostly been
targeted to specific cell types or organs by physicochemical targeting
in which LNP’s lipid compositions are adjusted to find mixtures with the
desired tropism. Here lung-tropic LNPs are examined, whose organ
tropism derives from containing either a cationic or ionizable lipid
conferring a positive zeta potential. Surprisingly, these LNPs are found
to induce massive thrombosis. Such thrombosis is shown in the lungs and
other organs, and it is shown that it is greatly exacerbated by
pre-existing inflammation. This clotting is induced by a variety of
formulations with cationic lipids, including LNPs and non-LNP
nanoparticles, and even by lung-tropic ionizable lipids that do not have
a permanent cationic charge. The mechanism depends on the LNPs binding
to and then changing the conformation of fibrinogen, which then
activates platelets and thrombin. Based on these mechanisms, multiple
solutions are engineered that enable positively charged LNPs to target
the lungs while ameliorating thrombosis. The findings illustrate how
physicochemical targeting approaches must be investigated early for
risks and re-engineered with a careful understanding of biological
mechanisms.
1 Introduction
Since the first Food and Drug Administration (FDA) approval
of a solid LNP in 2018, LNPs have rapidly become the preferred drug
delivery platform of the biopharma industry, with the trend accelerating
markedly after the development of the COVID-19 LNP vaccines.[1]
Targeting LNPs to specific organs or cell types is desirable for many
applications and is generally accomplished by one of two methods. The
older method is to conjugate LNPs to affinity moieties (e.g.,
antibodies) that bind to a known epitope on a target cell.[2]
This method presents major challenges for scale-up manufacturability
and immunogenicity, partially contributing to it never having produced
an FDA-approved targeted LNP or liposome. The second, and newer, method
for LNP targeting is to screen large numbers of LNP formulations for
those that have fortuitous “physicochemical tropism” to a target organ.
In physicochemical tropism/targeting, some (usually unknown) physical or
chemical features of the LNP cause it to enter into particular cells.
Over the last decade, the physicochemical approach has become the
dominant method of achieving targeting in both academia and industry,
because of the ease of in vivo screening and the highly efficient
manufacturing process. As physicochemical tropism has been able to
target many different organs and cell types, it has the potential to
meet LNPs’ promise of being able to modulate any protein in any cell
type or organ.
Physicochemical targeting is usually achieved by screening
many lipids, typically varying each of the four major lipid classes used
in LNPs: ionizable lipids, PEGylated lipids, cholesterol, and helper
lipids (phospholipids, sphingolipids, etc). The mechanisms of targeting
are rarely elucidated. As a notable exception, ApoE was implicated as
the plasma protein that binds to the first FDA-approved LNP, patisiran,
shuttling the LNPs to hepatocytes.[3, 4]
While mechanisms of delivery generally are not elucidated, there have
been some clear trends in physicochemical properties that correlate with
organ distribution. For example, intravenously injected (IV) LNPs
formulated with negatively charged lipids often accumulate in the spleen[5-9] and LNPs formulated with positively charged lipids usually accumulate in the lungs[5, 7, 9-12] as found independently by many labs and companies. These observations were, in a 2020 Nature Nanotechnology paper, distilled into a simpler and more elegant way of targeting LNPs.[5]
Instead of using particular ratios of hard-to-synthesize ionizable
lipids or exotic helper lipids, this seminal study showed it was
possible to add to a base formulation of LNPs either a negatively
charged lipid to target the spleen or a positively charged lipid to
target the lungs. This approach leads to democratization and
reproducibility since any lab or company can obtain such common lipids,
rather than having to synthesize the ionizable lipid variants used in a
particular lab's screening.
However, while testing such promising
physicochemically-targeted LNPs as therapeutics, we found that the
positively charged LNPs with lung tropism induce a major unreported side
effect: thrombosis. Studies of nanoparticle toxicities have largely
focused on two of the three major defense systems of plasma: complement
proteins and immunoglobulins.[13-15] However, the 3rd
defense system, clotting, has often been neglected in recent years,
even though it has the most deadly and acute consequences if improperly
activated. Blood has evolved to actively clot in response to different
foreign surfaces and the coagulation cascade is specifically known to
activate in response to charged surfaces. An IV dose of nanoparticles
exposes the blood to large amounts of foreign surface area, but
nanoparticles can also indirectly activate clotting by interacting with
cells that affect clotting processes, such as endothelial cells,
neutrophils, and platelets. Noting that the dominant nanomedicine
targeting method now employs manipulation of nanoparticle charge, it is
important to revisit the thrombotic side effects of nanomedicines[16-21] with a focus on the new targeting approaches.
Here, we show that lung tropic, positively charged, LNPs
induce massive thrombosis. This finding was quite general, as it held
for LNPs with different permanently positively charged lipids,
regardless of their co-loaded ionizable lipid, and not just LNPs, but
also liposomes. Perhaps most surprisingly, we also observed thrombosis
caused by LNPs that lacked a permanently cationic lipid (one possessing a
quaternary amine) but instead contained an ionizable lipid (possessing a
tertiary amine) that was found via screening to confer lung tropism.
The lung-tropic LNPs induced profound pulmonary embolism when
administered intravenously, induced stroke-like effects when
administered via the carotid artery, and modified clotting processes in
ex vivo blood. We investigated detailed mechanisms of LNP-induced
clotting, showing that lung-tropic LNPs bind to the core clotting
protein, fibrinogen, alter its secondary structure, and cause
aggregation and activation of platelets. Elucidating these detailed
mechanisms of clotting enabled us to develop three solutions that may
permit positively charged LNPs to maintain their lung-targeting property
while ameliorating dangerous clotting: Pre-treatment with
anticoagulants (direct thrombin inhibitors, but not the seemingly
obvious choice of heparin), conjugation of the LNPs to direct thrombin
inhibitors, or reduction of the LNP size.
2 Results
2.1 Lung-Tropic LNPs with Cationic Lipids Induce Gross Side Effects In Vivo
We fabricated physicochemically-targeted, lung-tropic
LNPs by the previously published method of adding in a cationic lipid,
1,2-dioleoyl-3-trimethylammonium-propane (DOTAP, chloride salt). We
initially synthesized these LNPs with the ionizable lipid cKK-E12
(herein referred to as +DOTAP LNPs, unless otherwise stated). This base
+DOTAP LNP formulation consisted of the lipids DOTAP, cKK-E12,
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE),
cholesterol, 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene
glycol-2000 (DMG-PEG 2000) (50/25/5/18.5/1.5, mol/mol) and mRNA
(lipid/mRNA ratio = 40/1, wt/wt). Control, non-lung tropic LNPs were
synthesized without DOTAP (Figure 1a).
There was no significant difference in the sizes of these LNPs (99.22 ±
4.27 nm for −DOTAP LNPs and 102.47 ± 0.48 nm for +DOTAP LNPs) (Figure 1b).
However, +DOTAP LNPs had a significantly higher zeta potential than
−DOTAP LNPs (14.34 mV vs −4.05 mV) due to the addition of the cationic
lipid (Figure 1c).
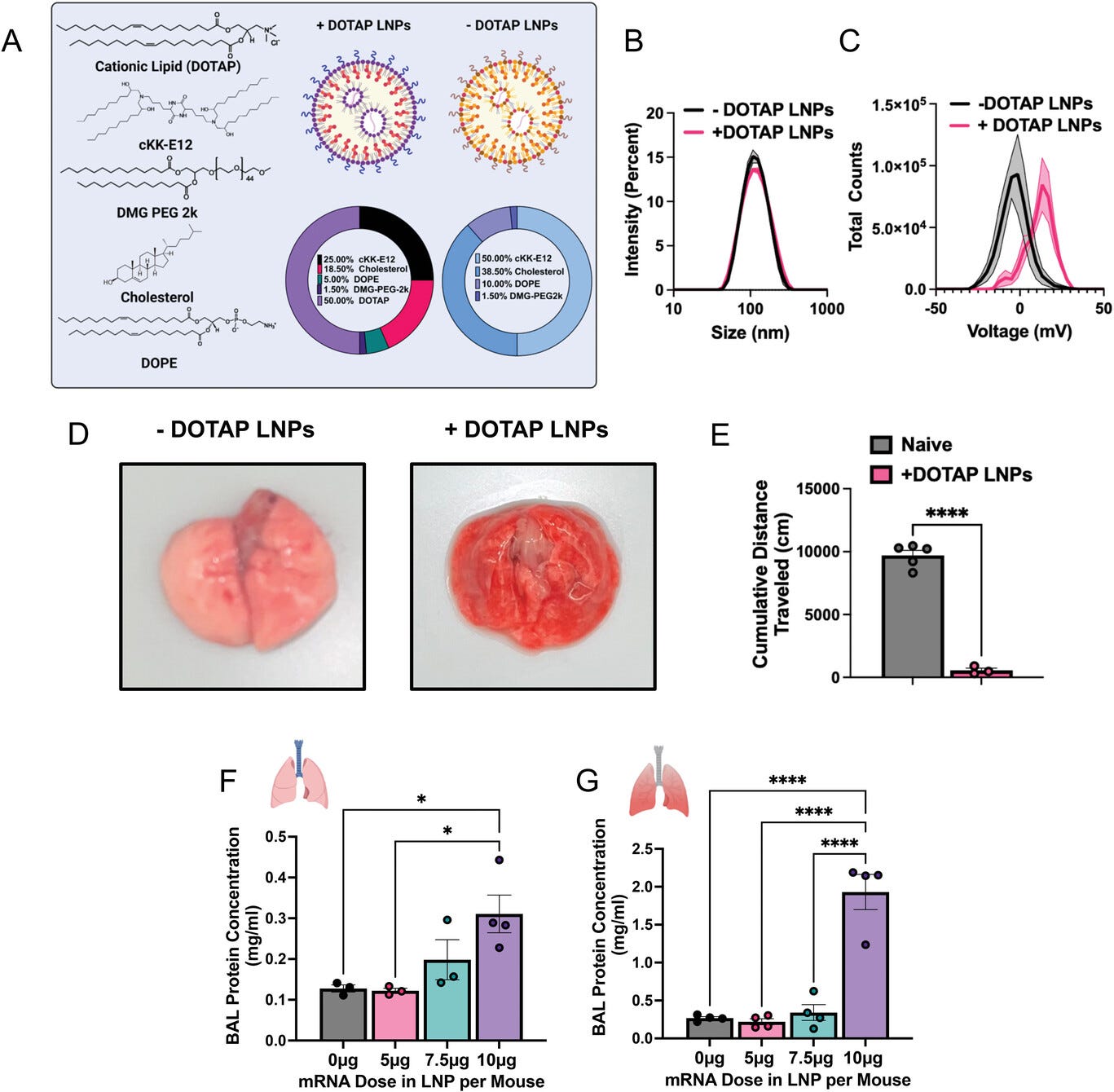
Figure 1
Open in figure viewerPowerPoint
Cationic LNPs with physicochemical tropism to the lungs induce acute, severe side effects in mice. A) Formulation of −DOTAP and +DOTAP LNPs. B) Size distribution of −DOTAP and +DOTAP LNPs determined by dynamic light scattering (DLS) shows no significant difference in size (99.22 ± 4.27 nm for −DOTAP LNPs and 102.47 ± 0.48 nm for +DOTAP LNPs). C) Zeta potential graphs of −DOTAP LNP (black) versus +DOTAP LNP (pink), show that −DOTAP LNPs are slightly negatively charged at −4.05 mV and +DOTAP LNPs have a surface charge of 14.34 mV. D) Gross anatomical comparison of naïve mice intravenously (IV) injected with either −DOTAP LNPs or +DOTAP LNPs reveals severe lung hepatization (liver-like appearance) with +DOTAP LNP injected mice. LNPs were allowed to circulate for 30 min. E) Comparison of cumulative distance traveled for 1 h after injection of +DOTAP LNPs assessed by AI software, DeepLabCut, compared to healthy, uninjected mice reveals a significant reduction in total distance traveled, indicating lethargy in these mice. F) Dose-response of the effect of +DOTAP LNP dose on protein concentration in bronchoalveolar lavage (BAL) fluid shows a dose-dependent increase in BAL protein, a measure of endothelial barrier dysfunction. G) This is further exacerbated in mice with pre-existing lung inflammation (nebulized LPS mice; note the 5-fold y-axis augmentation between (F) and (G)). Statistics: n = 3–5 and data shown represents mean ± SEM; For (E), comparisons between groups were made using an unpaired t-test with Welch's correction. For all other graphs, comparisons between groups were made using one-way ANOVA with Tukey's post-hoc test. * = p < 0.05, ** * = p < 0.001, **** = p < 0.0001.
We next intravenously (IV) injected the above LNPs into
naive mice at a dose of 10 µg of mRNA per mouse and sacrificed 30 min
later for gross anatomical inspection. We observed that in mice that
received +DOTAP LNPs, the lungs were strikingly red and firm, resembling
“hepatization” (liver-like appearance) described by pathologists for
human acute respiratory distress syndrome (ARDS) (Figure 1d). We observed clear lethargy and sluggishness in +DOTAP LNP mice. Using the software DeepLabCut as previously described,[22]
we found that after a recording time interval of 1 h post-LNP
injection, +DOTAP LNP mice walked a total distance that was > 17-fold
less than naïve mice (Figure 1e).
We further assessed lung-specific toxicity by measuring the contents of
the bronchoalveolar lavage (BAL) fluid 20 h after IV +DOTAP LNP
injection, which assesses lung capillary leak. In naive mice, there was a
dose-dependent increase in both BAL protein and leukocyte count from 5
to 10 µg of mRNA (Figure 1f and Figure S1, Supporting Information).
Because lung-targeted LNPs will generally be given to
patients with lung pathology, we next investigated whether +DOTAP LNPs
caused even more severe injury in mice with pre-existing lung
inflammation. We utilized a mouse model of acute lung inflammation
achieved by administering nebulized lipopolysaccharides (LPS). +DOTAP
LNPs were IV injected 4 h after LPS exposure and mice were sacrificed 20
h later. As in naïve mice, we saw a dose-dependent increase in BAL
protein concentration from 5 to 10 µg of mRNA (Figure 1g).
However, in nebulized LPS mice, there was an exacerbation
of BAL edema at each dose tested. At the 10 µg dose of mRNA, nebulized
LPS mice have 6-fold higher BAL edema compared to naïve mice injected
with +DOTAP LNPs. This indicates that the toxicity of +DOTAP LNPs is
amplified under pre-existing inflammatory conditions. Based on these
data, a dose of 10 µg of mRNA in LNPs was used for further in vivo
studies unless otherwise stated. This is a therapeutically relevant dose
for mRNA-LNP delivery and enabled us to adequately assess the
mechanisms behind DOTAP LNP toxicity.
2.2 Coagulation is Triggered by LNPs and Other Nanoparticles with Cationic Lipids, across Diverse Formulations
Given the very acute nature of the side effects of +DOTAP
LNPs, seen within a minute on the AI-measured distance-walked test, we
hypothesized that the nanoparticles were activating a protein-based
defense system in the blood. The three major plasma protein defense
systems are the complement cascade, immunoglobulins, and the coagulation
cascade. Since complement has been long implicated in nanoparticle
defense,[13-15]
and since immunoglobulins’ acute effects also lead to complement
activation (via IgG-mediated activation of the classical pathway of
complement), we first investigated if these LNPs activate the complement
system. Surprisingly, we discovered that +DOTAP LNPs do not activate
complement, as we detected no increase in the plasma concentration of
C3a 30 min after +DOTAP LNP IV injection into naive mice (Figure S2, Supporting Information), noting that we have extensively shown C3a production induced by other nanoparticles[13, 14]
Without C3a production, it is highly unlikely that complement
activation, or acute immunoglobulin effects, mediate +DOTAP LNPs’ acute
toxicity.
Therefore, we investigated whether the toxicity was
produced by the coagulation cascade. To investigate this, we stained
lung sections with Masson's trichrome to look for evidence of clots.
Lung histological samples show large clots both at the large vessel and
capillary levels in naïve mice IV injected with +DOTAP LNPs, compared to
mice injected with −DOTAP LNPs where there are no observable clots (Figure 2a).
This was a clear indication of thrombosis (clotting) due to +DOTAP
LNPs. In these mice, visible clots were not observed in the liver or the
spleen (Figure S3a,b,
Supporting Information). However, this effect is not restricted to the
lungs since we also observed evidence of clotting in the brain when
+DOTAP LNPs were injected via the carotid artery (Figure S3c,
Supporting Information). These results indicate the incidence of clots
in the first capillary bed downstream of injection (lungs after IV
injection; brain after intra-arterial injection). To quantify the extent
of clotting, we measured thrombin-antithrombin (TAT) plasma levels in
both naïve and nebulized-LPS injured mice. TAT is a stable complex
formed after thrombin activation that serves as a marker of coagulation.
We found that +DOTAP LNPs increase TAT levels by > 2-fold in naive
mice and by an additional > 1.5-fold in nebulized-LPS injured mice
(Figure 2b).
This confirmed that +DOTAP LNPs activate thrombin (the common pathway
of coagulation), and this is exacerbated in pre-existing inflammatory
conditions, probably as a result of thrombo-inflammation.[23]
Furthermore, we show that in addition to permanently cationic lipids
like DOTAP, ionizable cationic lipids that are used to induce lung
tropism also induce clotting. An example of such an ionizable lipid is
306-N16B, which was shown in a recent PNAS paper to produce excellent
lung tropism[24] and is also cationic at neutral pH (Figure S4 and Table S1,
Supporting Information). However, we found that 306-N16B LNPs also
increase TAT plasma levels by ≈2.5-fold in naïve mice (Figure 2c) and demonstrate other indications of coagulation (Figure S5,
Supporting Information). This finding therefore generalizes this
clotting phenomenon to a wide variety of physicochemically-targeted LNPs
that rely on positive charge for lung localization.
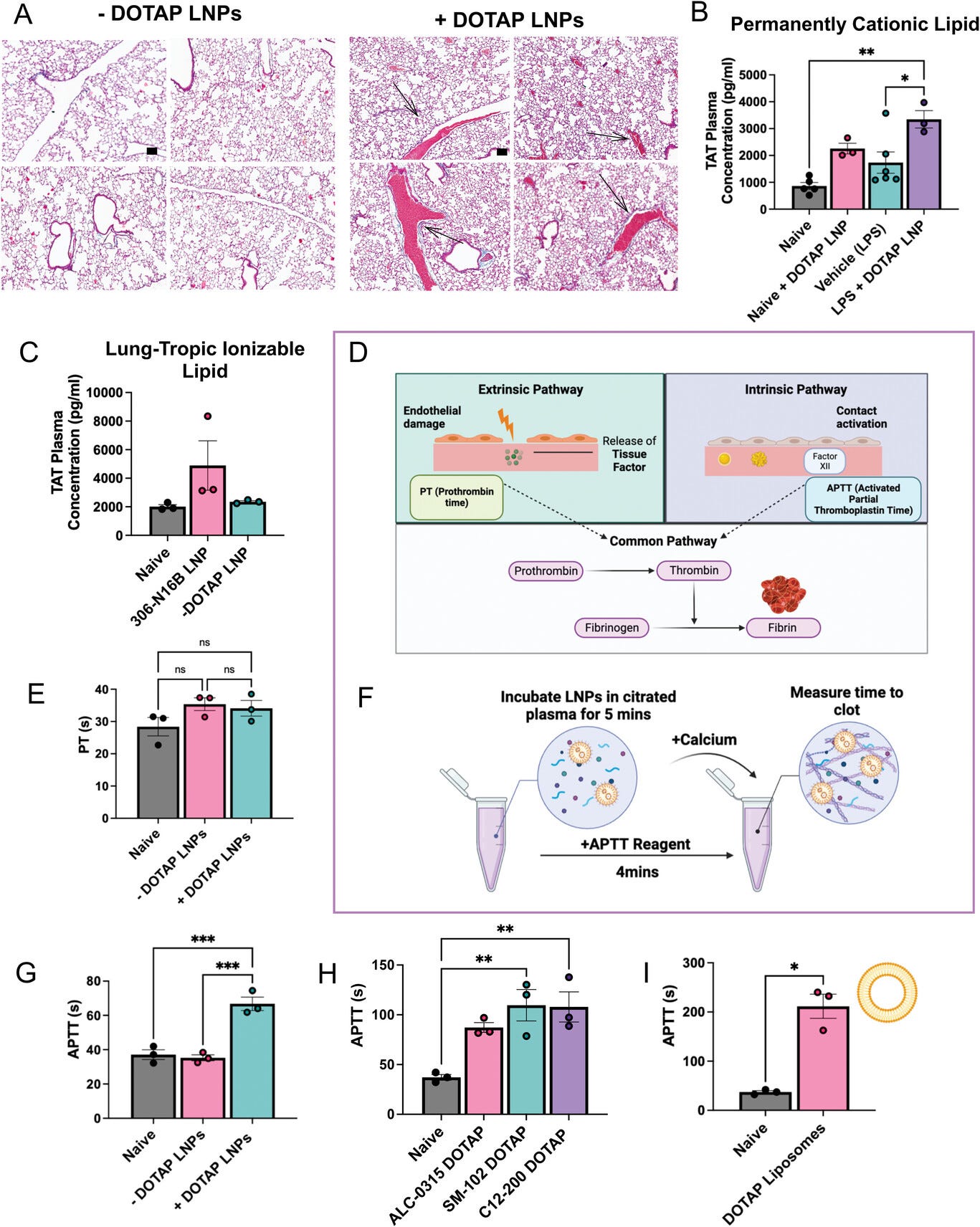
Figure 2
Open in figure viewerPowerPoint
Coagulation is activated by positively charged LNPs and other nanoparticles, across diverse formulations, both in vivo and in ex vivo whole blood. A) +DOTAP LNPs induce large clots (black arrows) in the pulmonary arteries (scale bar = 100 µm). −DOTAP or +DOTAP LNPs were injected into healthy mice at a dose of 10 µg of mRNA and 30 min after, lungs were harvested and prepared for histology (Masson's trichrome). −DOTAP LNPs do not show such clots. B) In vivo, +DOTAP LNPs increase plasma levels of thrombin-antithrombin (TAT), a marker of recent clotting. The TAT increase is > 2-fold in naïve mice, with an additional > 1.5-fold increase in mice with pre-existing lung inflammation (nebulized-LPS). +DOTAP LNPs were injected into naïve mice and 30 min post-injection, plasma was collected for TAT assay. C) 306-N16B LNPs increase TAT plasma levels by ≈2.5-fold compared to naive mice while control −DOTAP LNPs do not. 2E11 LNPs per mouse were injected and TAT plasma levels were measured after 30 min. D) Schematic of the two major coagulation pathways – intrinsic and extrinsic – which both converge into the common pathway. The extrinsic pathway is triggered by damage that occurs outside the blood vessel, exposing tissue factors. The intrinsic pathway is initiated by factors within the blood vessel lumen. Both the intrinsic and extrinsic pathways converge to the common pathway which involves the conversion of prothrombin to thrombin – an enzyme that converts fibrinogen to fibrin. Fibrin forms the mesh-like framework of clots. The prothrombin time (PT) and activated partial thromboplastin time (APTT) are laboratory tests used to measure the time to clot through the extrinsic and intrinsic coagulation pathways, respectively. E) +DOTAP LNPs do not change PT time in vitro, showing that they do not induce coagulation through the extrinsic coagulation pathway. F) Timeline and schematic of in vitro APTT measurements. G) +DOTAP LNPs increase APTT, showing that they induce coagulation through the extrinsic pathway. H) +DOTAP LNPs increase APTT through the intrinsic pathway regardless of ionizable lipid identity, as ALC-0315, SM-102, and C12-200 DOTAP LNPs also show elevated APTT. I) Positively charged, non-LNP nanoparticles, here liposomes containing DOTAP, also increase APTT time. Statistics: n = 3–6 and the data shown represents mean ± SEM; For (I), comparisons between groups were made using an unpaired t-test with Welch's correction. For all other graphs, comparisons between groups were made using one-way ANOVA with Tukey's post-hoc test. * = p < 0.05, *** = p < 0.001, **** = p < 0.0001.
We sought to determine which molecular coagulation
pathways underlie LNP-induced thrombosis. Coagulation can be divided
into the intrinsic and extrinsic pathways (Figure 2d).
The extrinsic pathway is activated by trauma that damages the
endothelium, leading to exposure of tissue factor which activates the
coagulation protein factor VII. The intrinsic pathway is initiated by
factors within the blood vessel lumen and has been shown to be triggered
by charged surfaces (such as foreign particles) or collagen, which
trigger the activation of the coagulation protein factor XII.[25]
The extrinsic and intrinsic pathways converge to the common pathway
which involves the conversion of prothrombin to thrombin – the enzyme
that converts fibrinogen to fibrin. Fibrin forms the mesh-like framework
of clots. The prothrombin time (PT) and activated partial
thromboplastin time (APTT) are laboratory tests used to measure the time
to clot through either the extrinsic or intrinsic pathways,
respectively. To determine if +DOTAP LNP-activated coagulation employs
the intrinsic or extrinsic pathway, we measured PT and APTT after doping
LNPs into plasma in vitro. While +DOTAP LNPs did not change the PT
(Figure 2e), they led to a significant increase in APTT (Figure 2f,g).
This indicates that +DOTAP LNPs activate the intrinsic coagulation
pathway, thus depleting intrinsic pathway proteins and prolonging the
APTT. Furthermore, the effects of +DOTAP LNPs on coagulation are
dependent on both the number of LNPs injected and the mole fraction of
DOTAP in the formulation (Figure S6b,d,e, Supporting Information).
We formulated liposomes and other LNPs with DOTAP to see
if the coagulation side effects were a generalizable result of
co-formulation with cationic lipids. We formulated LNPs with various
ionizable lipids (C12-200, ALC-0315, SM-102; Figure S4 and Table S1, Supporting Information) and found that +DOTAP LNPs increase APTT, regardless of the ionizable lipid type (Figure 2h). We used DOTMA, rather than DOTAP, as our cationic lipid and observed the same effect on APTT (Figure S7, Supporting Information). Adding DOTAP to liposomes, rather than LNPs, also results in increased APTT (Figure 2i and Figure S8a, Supporting Information). Finally, 306-N16B LNPs also lead to an increase in clotting time in vivo (Figure S5d,
Supporting Information). Thus, this clotting phenomenon is
generalizable to different LNP and liposome formulations containing both
ionizable and permanently cationic lipids. Notably, these side effects
appear to be associated with a positive charge as anionic LNPs
formulated with the negatively charged lipid DOPS do not induce clotting
(Figure S8b,c, Supporting Information).
2.3 LNPs with Cationic Lipids Aggregate and Activate Platelets
Having proven that +DOTAP LNPs activate the coagulation
cascade, we sought to assess if +DOTAP LNPs also cause activation and
aggregation of platelets, since clots can also form due to platelet
aggregates.[26]
We measured complete blood counts (CBCs) in blood drawn 30 min
post-injection of +DOTAP LNPs. CBCs showed that +DOTAP LNPs greatly
reduced the number of circulating platelets in mice with pre-existing
inflammation (nebulized LPS), and also showed a trend towards reduced
platelets in naive mice (Figure 3a).
This reduction in platelet count is dependent on both the number of
+DOTAP LNPs injected and the mol fraction of DOTAP in the LNP
formulation (Figure S6a,c,
Supporting Information). Notably, LNPs targeted to the lung using
antibodies against platelet endothelial cell adhesion molecule (PECAM)
do not have the same effect on platelets showing that this is due to a
positive charge (Figure S9,
Supporting Information). Furthermore, ionizable cationic 306-N16B LNPs
also significantly decrease platelet count when injected into naïve mice
(Figure 3b).
Reduced platelet count (thrombocytopenia) can indicate depletion of
platelets from circulation due to incorporation in clots. We wanted to
investigate if this thrombocytopenia was due to platelet activation by
+DOTAP LNPs. To measure platelet activation, we employed flow cytometry.
As shown in the schematic of Figure 3c,
we used forward scatter (FSC) and side scatter (SSC) measurements to
detect larger and more complex platelet aggregates versus small,
isolated platelets. We stained for two specific markers of platelets:
Glycoprotein V (GPV; CD42d), and P-selectin (CD62p) (Figure S10a,d, Supporting Information). CD42d is constitutively found on the surface of all platelets.[27]
In resting platelets, P-selectin is generally stored in platelet
granules, which are externalized when the platelet becomes activated.[27, 28] Platelet-rich plasma (PRP; Figure 3d) was either left untreated or incubated with fluorescently labeled −DOTAP or +DOTAP LNPs and stained for CD42d and P-selectin.
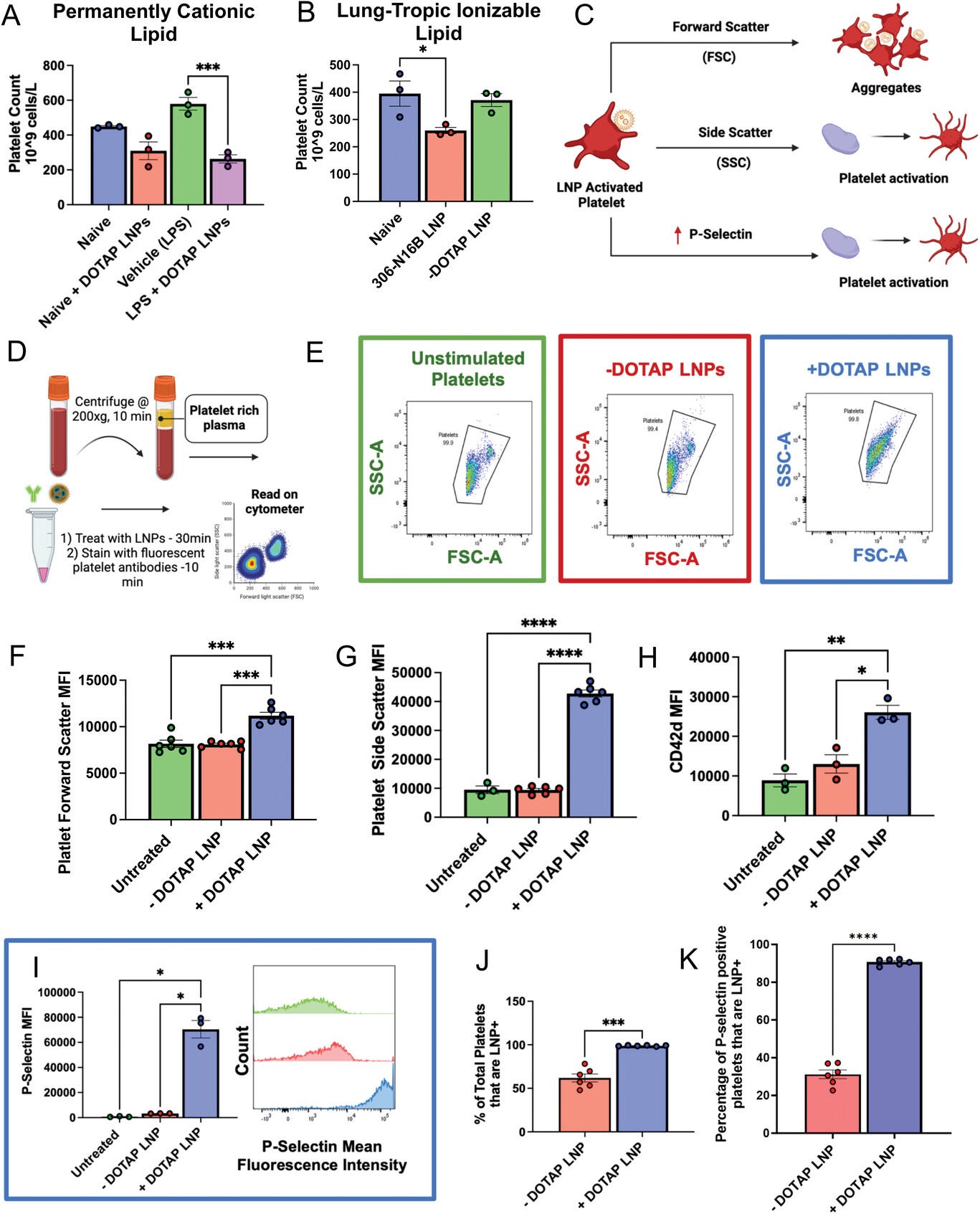
Figure 3
Open in figure viewerPowerPoint
DOTAP LNPs cause platelet activation. A) +DOTAP LNPs significantly decrease platelet count in naive mice and even more so in mice with pre-existing inflammation (nebulized LPS). LNPs were IV-injected and platelet count was measured from whole blood 30 min later. B) 306-N16B LNPs decrease platelet count in naïve mice while control −DOTAP LNPs do not. C) Schematic depicting what different flow cytometry metrics describe platelet physiology. D) Schematic of protocol used for platelet flow cytometry. After isolation of platelet-rich plasma (PRP), samples were either left untreated or treated with −DOTAP or +DOTAP LNPs for an incubation period of 30 min at 37 °C. E) Representative FSC-A versus SSC-A graphs of unstimulated platelets (green) and platelets treated with +DOTAP (blue) or −DOTAP LNPs (red), showing a clear change in the scatterplot with +DOTAP LNPs. There are significant increases in F) FSC and G) SSC upon treatment with +DOTAP LNPs suggesting larger and more complex platelet aggregates. +DOTAP LNPs cause upregulation of H) CD42d and I) P-selectin, based on mean fluorescence intensity (MFI). J) Platelets were assayed for whether they were physically associated with LNPs, as measured by the fraction of platelets that were positive for a fluorescent lipid that had been incorporated into the LNPs during synthesis. While 50% of platelets were associated with −DOTAP LNPs, nearly 100% of platelets were physically associated with +DOTAP LNPs. K) Staining with P-selectin shows that while only 30% of P-selectin positive platelets were associated with −DOTAP LNPs, almost 100% of P-selectin positive platelets were LNP positive as well, showing preference for an activated state. Statistics: n = 3–6 and data shown represents mean ± SEM; For (J,K), Welch's t-test was performed. For all other graphs, Brown-Forsythe and Welch's ANOVA was performed with the Dunnett T3 post-hoc test. * = p < 0.05, *** = p < 0.001, **** = p < 0.0001.
FSC versus SSC plots for platelets (CD42d-positive
events) in untreated, −DOTAP, and +DOTAP LNP-treated PRP show no
difference between the untreated and −DOTAP LNP-treated conditions, but
the shape of the +DOTAP LNP graph differs markedly from the other two
(Figure 3e).
The mean FSC and SSC values are elevated for platelets in the presence
of +DOTAP LNPs, indicating the formation of aggregates (Figure 3f,g).
Further evidence of platelet aggregation induced by +DOTAP LNPs is
shown by increased CD42d signal for each CD42d-positive event (Figure 3h).
In the presence of +DOTAP LNPs, each CD42d-positive event is a cluster
of platelets, rather than an individual cell, so the mean CD42d signal
is increased versus untreated and −DOTAP LNP-treated samples. Most
interesting, however, is the 20-fold increase in P-selectin presentation
induced by +DOTAP LNPs (Figure 3i).
Since increased surface presentation of P-selectin is a marker of
platelet activation, this demonstrates profound platelet activation
induced by +DOTAP LNPs.
Tracing LNP fluorescence in our flow cytometry
measurements, we found that 100% of platelets were physically associated
with fluorescently labeled +DOTAP LNPs, while only 50% of platelets
were physically associated with −DOTAP LNPs, showing that the cationic
lipid profoundly increases LNP adhesion to platelets (Figure 3j).
Specifically, among platelets that had an LNP signal, we assessed the
P-selectin signal. For −DOTAP-LNPs, only 30% of the LNP-associated
platelets were P-selectin positive, indicating that −DOTAP LNPs had
little effect on platelet activation. However, for +DOTAP LNPs, nearly
100% of LNP-positive platelets were also positive for P-selectin,
indicating that platelet association with +DOTAP LNPs predicts platelet
activation (Figure 3k).
2.4 DOTAP LNPs bind to Fibrinogen, and Fibrinogen is Required for LNP-Induced Platelet Activation
Having proven that +DOTAP LNPs cause clotting, initiate
the coagulation cascade, and activate platelets, we sought to isolate
components of the +DOTAP LNP protein corona that might lead to these
effects. Upon IV injection, LNPs can form unique protein coronas based
on their physicochemical properties and this could alter their
functionality.[29, 30]
The protein corona can therefore drive the biodistribution and
cell-type localization of LNPs. However, the proteins adsorbed on LNPs
could also induce side effects. Proteins can undergo conformational
changes when adhering to surfaces and aggregates of proteins on surfaces
do not often behave similarly to proteins in solution, including
causing immunogenic or thrombotic side effects.[29]
We therefore hypothesized that +DOTAP LNPs could bind to
coagulation proteins in plasma, the most abundant being fibrinogen.
Fibrinogen is a soluble protein found in plasma. When it is cleaved by
thrombin, fibrinogen undergoes conformational changes and aggregates to
form fibrin strands, which in turn form the clot-stabilizing mesh.
Fibrinogen/fibrin aggregates on surfaces could cause adhesion and
activation of platelets. Fibrinogen has a net negative charge at
physiological pH and contains domains with a high concentration of
negatively charged residues, such as the E domain.[31, 32] Studies have linked the exposure of certain fibrinogen residues to specific side effects.[33-35]
For example, nanoparticle-induced exposure of a peptide sequence at the
C-terminus of the fibrinogen γ-chain interacts with the integrin
receptor Mac-1 and induces inflammation.[35]
We therefore hypothesized that the positively charged DOTAP in LNPs
could interact with, and aggregate fibrinogen in a way that could lead
to coagulation and platelet activation. This hypothesis is supported by
our finding that +DOTAP LNPs decrease the expression of CD41 on
platelets (Figure S10b,
Supporting Information). Since CD41 is the platelet receptor of
fibrinogen/fibrin, this indicates that there is a blockage of this
marker by fibrinogen induced by +DOTAP LNPs.
We first tested if +DOTAP LNPs bind to fibrinogen in
vitro. We added fluorescently labeled fibrinogen and LNPs to the plasma
and incubated for 10 min. We diluted this sample and performed
fluorescence-mode nanoparticle tracking analysis (NTA) to detect
nanoparticle-sized aggregates of fluorescent fibrinogen, indicating
fibrinogen aggregation on LNPs (Figure 4a).
Approximately 38-fold more fibrinogen-positive particles were observed
in the presence of +DOTAP LNPs versus −DOTAP LNPs (Figure 4b–d).
This agrees with our findings that +DOTAP LNPs aggregate significantly
with fibrinogen in plasma and even after incubation with fibrinogen
alone (Figures S11 and S12, Supporting Information).
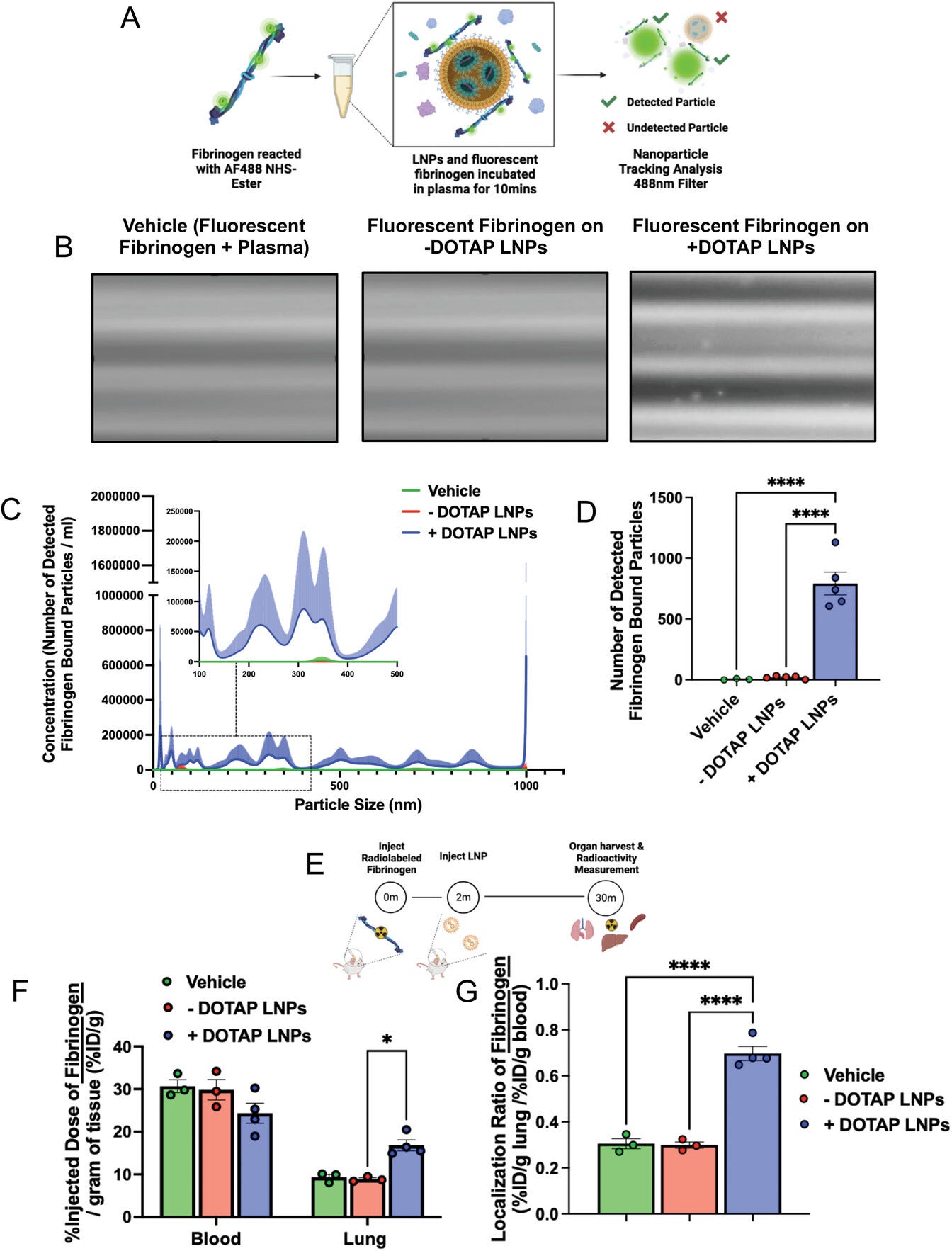
Figure 4
Open in figure viewerPowerPoint
Fibrinogen binds to +DOTAP LNPs, is required for LNP-induced activation of platelets, and is likely the first step in LNP-induced clotting. A) To measure LNP binding to fibrinogen, we fluorescently labeled fibrinogen, mixed it with plasma and LNPs, and used nanoparticle tracking analysis (NTA) to identify individual nanoparticles that had become bound to fluorescent fibrinogen. B) Representative NTA images of fluorescent fibrinogen on −DOTAP or +DOTAP LNPs, showing visible binding of fibrinogen to +DOTAP LNPs but no visible binding to −DOTAP LNPs, or vehicle control. C) NTA data was turned into particle size versus concentration histograms, and here we plot fibrinogen-positive particles only. +DOTAP LNPs bind strongly to fluorescent fibrinogen and this leads to LNPs of many different sizes, while −DOTAP LNPs barely have any fibrinogen-positive signal at any particle size. Inset: Size versus concentration histogram ranging from 100–500 nm. D) Number of detected fibrinogen-bound particles from (C) showing that +DOTAP LNPs generate ≈37.7× more fibrinogen-bound particles than −DOTAP LNPs. E) Next, we IV-injected radiolabeled fibrinogen into mice, followed by LNPs 2 min later. Organs were harvested 30 min after LNP injection and the radioactivity in each organ was measured. F) Amount of radiolabeled fibrinogen in the blood and lungs after the protocol of (E), shown as the % of injected dose per gram of tissue (%ID g−1). +DOTAP LNPs lead to a ≈2× increase in fibrinogen lung uptake, while −DOTAP LNPs do not alter fibrinogen biodistribution compared to control. G) Localization ratios of fibrinogen from (F) calculated as %ID g−1 of tissue in the lung divided by that in the blood. +DOTAP LNPs cause a 2.4-fold higher localization ratio of fibrinogen compared to −DOTAP LNPs and vehicles. H) Representative confocal microscopy images of lung sections from mice injected with fluorescent fibrinogen followed by fluorescent LNPs, showing visible fibrinogen aggregates in the lungs of +DOTAP LNP injected mice (n = 3 biologically independent animals). I) Mean fluorescence intensities (MFIs) in the lung sections from (H) of − or +DOTAP LNPs and fibrinogen showing significantly higher MFIs of LNPs and fibrinogen in +DOTAP LNP injected mice compared to −DOTAP LNP mice. J) From the microscopy images in (H), the percentage of fibrinogen signal overlap with that of LNPs or percentage of LNP signal overlap with fibrinogen signal in the presence of − or +DOTAP LNPs shows a strong co-localization of fibrinogen with +DOTAP LNPs. K) Circular dichroism of fibrinogen (1 mg ml−1) in the presence of −DOTAP or +DOTAP LNPs (4E11 LNPs per ml) reveals that +DOTAP LNPs alter the secondary structure of fibrinogen while −DOTAP LNPs do not. L) To determine if fibrinogen is necessary for LNP-induced activation of platelets, we added tPA to plasma to deplete the fibrinogen. Fibrinogen depletion nearly completely prevented LNP-induced activation of platelets. Statistics: n = 4–12 for (I) and (J) and n = 3–4 for all other graphs. Data shown represents mean ± SEM; For (F), (I), and (J), comparisons between groups were made using two-way ANOVA with Tukey's post-hoc test. For all other graphs, comparisons between groups were made using one-way ANOVA with Tukey's post-hoc test. * = p < 0.05, *** = p < 0.001, **** = p < 0.0001.
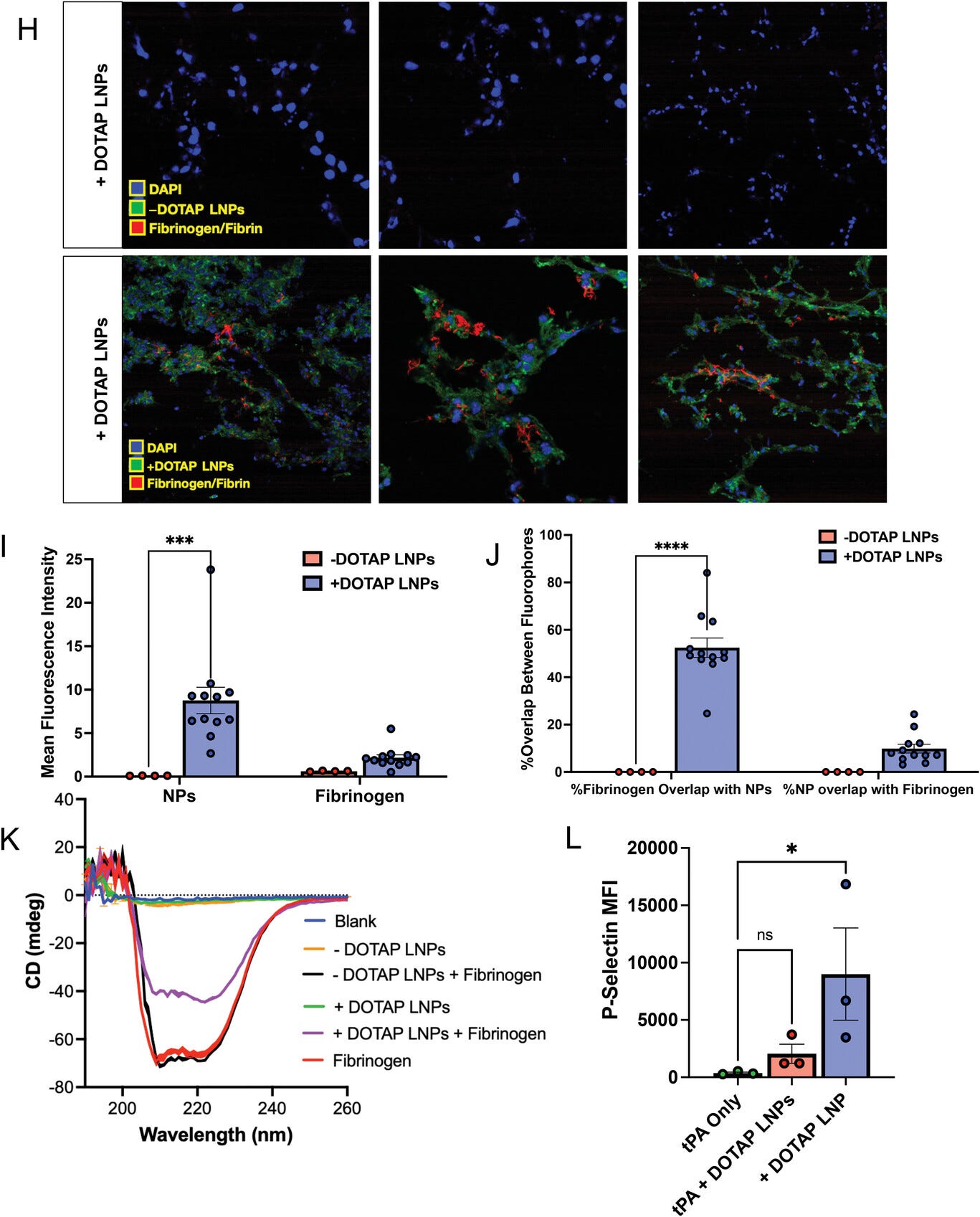
Figure 4 (continued)
Open in figure viewerPowerPoint
Fibrinogen binds to +DOTAP LNPs, is required for LNP-induced activation of platelets, and is likely the first step in LNP-induced clotting. A) To measure LNP binding to fibrinogen, we fluorescently labeled fibrinogen, mixed it with plasma and LNPs, and used nanoparticle tracking analysis (NTA) to identify individual nanoparticles that had become bound to fluorescent fibrinogen. B) Representative NTA images of fluorescent fibrinogen on −DOTAP or +DOTAP LNPs, showing visible binding of fibrinogen to +DOTAP LNPs but no visible binding to −DOTAP LNPs, or vehicle control. C) NTA data was turned into particle size versus concentration histograms, and here we plot fibrinogen-positive particles only. +DOTAP LNPs bind strongly to fluorescent fibrinogen and this leads to LNPs of many different sizes, while −DOTAP LNPs barely have any fibrinogen-positive signal at any particle size. Inset: Size versus concentration histogram ranging from 100–500 nm. D) Number of detected fibrinogen-bound particles from (C) showing that +DOTAP LNPs generate ≈37.7× more fibrinogen-bound particles than −DOTAP LNPs. E) Next, we IV-injected radiolabeled fibrinogen into mice, followed by LNPs 2 min later. Organs were harvested 30 min after LNP injection and the radioactivity in each organ was measured. F) Amount of radiolabeled fibrinogen in the blood and lungs after the protocol of (E), shown as the % of injected dose per gram of tissue (%ID g−1). +DOTAP LNPs lead to a ≈2× increase in fibrinogen lung uptake, while −DOTAP LNPs do not alter fibrinogen biodistribution compared to control. G) Localization ratios of fibrinogen from (F) calculated as %ID g−1 of tissue in the lung divided by that in the blood. +DOTAP LNPs cause a 2.4-fold higher localization ratio of fibrinogen compared to −DOTAP LNPs and vehicles. H) Representative confocal microscopy images of lung sections from mice injected with fluorescent fibrinogen followed by fluorescent LNPs, showing visible fibrinogen aggregates in the lungs of +DOTAP LNP injected mice (n = 3 biologically independent animals). I) Mean fluorescence intensities (MFIs) in the lung sections from (H) of − or +DOTAP LNPs and fibrinogen showing significantly higher MFIs of LNPs and fibrinogen in +DOTAP LNP injected mice compared to −DOTAP LNP mice. J) From the microscopy images in (H), the percentage of fibrinogen signal overlap with that of LNPs or percentage of LNP signal overlap with fibrinogen signal in the presence of − or +DOTAP LNPs shows a strong co-localization of fibrinogen with +DOTAP LNPs. K) Circular dichroism of fibrinogen (1 mg ml−1) in the presence of −DOTAP or +DOTAP LNPs (4E11 LNPs per ml) reveals that +DOTAP LNPs alter the secondary structure of fibrinogen while −DOTAP LNPs do not. L) To determine if fibrinogen is necessary for LNP-induced activation of platelets, we added tPA to plasma to deplete the fibrinogen. Fibrinogen depletion nearly completely prevented LNP-induced activation of platelets. Statistics: n = 4–12 for (I) and (J) and n = 3–4 for all other graphs. Data shown represents mean ± SEM; For (F), (I), and (J), comparisons between groups were made using two-way ANOVA with Tukey's post-hoc test. For all other graphs, comparisons between groups were made using one-way ANOVA with Tukey's post-hoc test. * = p < 0.05, *** = p < 0.001, **** = p < 0.0001.
We further sought to determine if +DOTAP LNPs bind to
fibrinogen in vivo and alter its biodistribution. Fibrinogen was
radioactively labeled and injected into mice, followed by + or DOTAP
LNPs 2 min later. Organs were harvested 30 min after LNP injection and
the radioactivity in each organ was measured (Figure 4e).
While −DOTAP LNPs do not alter fibrinogen biodistribution, there is a
≈2-fold increase in fibrinogen lung uptake in the presence of +DOTAP
LNPs (Figure 4f,g and Figure S13a,
Supporting Information). A similar +DOTAP LNP-induced increase in lung
fibrinogen uptake was observed in mice with pre-existing inflammation
(Figure S13b,
Supporting Information). Notably, the localization of fibrinogen to the
lungs induced by +DOTAP LNPs persists even 4 h after injection with
evidence of fibrinolysis only 24 h after (Figure S13c–f,
Supporting Information). Confocal microscopy images of lung sections
show visible aggregates of fibrinogen in +DOTAP LNP-injected mice and no
visible fibrinogen in −DOTAP LNP-injected mice (Figure 4h).
This was quantified across multiple images, with the LNPs and
fibrinogen having much more signal in the lungs of +DOTAP versus −DOTAP
LNP mice (Figure 4i).
Furthermore, fibrinogen is strongly associated with +DOTAP LNPs as
shown by the percentage of fibrinogen signal overlap with that of LNPs
or the percentage of LNP signal overlap with fibrinogen signal (Figure 4j). The data in Figure 4a–j
show that fibrinogen binds to +DOTAP LNPs (but not −DOTAP LNPs), and
this binding correlates with clotting in the lungs. We then performed
circular dichroism (CD) to determine if the binding of +DOTAP LNPs to
fibrinogen induces changes in the protein's secondary structure. The CD
spectra for fibrinogen alone show two negative peaks at 209 and 220 nm
which is characteristic of a protein with an α-helix (Figure 4k).
However, the addition of +DOTAP LNPs significantly reduces the
ellipticity of fibrinogen at both 209 and 220 nm while −DOTAP LNPs do
not. This indicates that the secondary structure of fibrinogen is
significantly altered in the presence of +DOTAP LNPs which supports the
hypothesis that this altered fibrinogen structure could induce platelet
activation and coagulation.
To further examine the effects of fibrinogen binding to
+DOTAP LNPs, we depleted fibrinogen from plasma doped with +DOTAP LNPs.
Since fibrinogen depletion would prevent coagulation, we checked if
fibrinogen depletion also abrogated +DOTAP LNP-induced platelet
activation. PRP was treated with tissue plasminogen activator (tPA)
which converts plasminogen to plasmin, causing degradation of
fibrinogen. We found that in fibrinogen-depleted plasma, +DOTAP LNPs do
not induce significant platelet activation as measured by P-Selectin MFI
(Figure 4l).
Thus, fibrinogen is necessary for +DOTAP LNP-induced platelet
activation. This fits with a mechanism in which positively charged LNPs
first bind to fibrinogen, inducing a conformational change in the
protein that activates platelets, which in turn activates the rest of
the coagulation cascade. This fits with prior studies that show that
immobilization of fibrinogen on surfaces can expose normally concealed
domains that activate platelets.[36-39]
2.5 Anticoagulation Ameliorates Cationic LNP-Induced Clotting
Below, we propose and test ways to prevent LNP-induced
clotting. To determine whether anticoagulation can reduce the toxicity
of +DOTAP LNPs, we compared clot formation between mice treated with
+DOTAP LNPs alone and those pre-treated with the clinically used
anticoagulants heparin and bivalirudin. Heparin functions by enhancing
the activity of endogenous antithrombin while bivalirudin is a direct
thrombin inhibitor. Anticoagulants were IV injected 5 min prior to
+DOTAP LNP injection (62.5U per mouse for heparin and 400 µg per mouse
for bivalirudin) and LNPs were allowed to circulate for 30 min. We found
that both heparin and bivalirudin pre-treatment significantly reduce
the formation of clots in the lung compared to +DOTAP LNP injection
alone, as shown by lung histological samples (Figure 5a).
We then investigated the effect of anticoagulation on +DOTAP LNP
biodistribution and mRNA expression. We radiolabeled LNPs with
indium-111 for biodistributions and injected mice with LNPs loaded with
luciferase mRNA to trace mRNA expression (Figure 5b).
Surprisingly, heparin pre-treatment shunted LNP localization from the
lung to the liver and spleen and completely attenuated luciferase
expression in the lungs (Figure 5c,d, and Figure S14a,
Supporting Information). We hypothesize that this unexpected effect is
because heparin is a negatively charged polysaccharide, so it could have
a charge interaction with +DOTAP LNPs and this could inhibit LNP
functionality, though other mechanisms may exist.[40]
By contrast, bivalirudin pre-treatment preserves the localization of
+DOTAP LNPs to the lung, as well as the LNP-induced mRNA luciferase
expression (Figure 5b,c, and Figure S14b,
Supporting Information). These studies illustrate that while the side
effects of +DOTAP LNPs can be ameliorated with anticoagulation, the
properties of each anticoagulant must be thoroughly examined to ensure
that LNP activity is preserved.
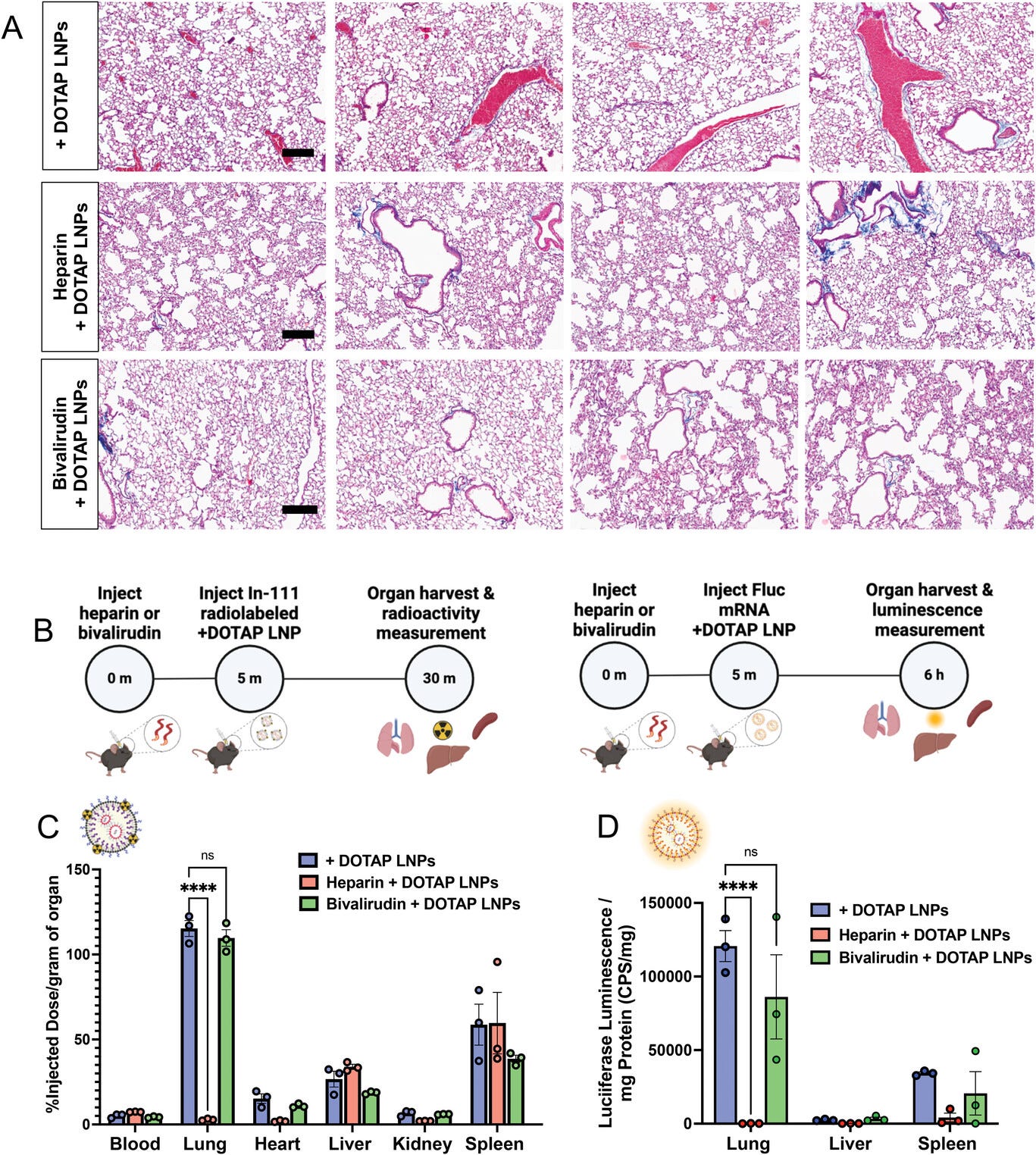
Figure 5
Open in figure viewerPowerPoint
Anticoagulation ameliorates the side effects of DOTAP LNPs, but only select anticoagulants still allow LNP targeting to the lungs. A) Histology showing clots in the lungs of mice given +DOTAP LNPs (top row), but not in mice that were given +DOTAP LNPs preceded by heparin (middle row) or bivalirudin (bottom row; scale bar = 200 µm). B) Schematics showing the protocol for biodistribution studies (using In-111-labeled LNPs) and luciferase expression studies. C) 30-min biodistribution in naïve mice of +DOTAP LNPs with no treatment or pretreated with heparin or bivalirudin. Pretreatment with heparin ablates localization of +DOTAP LNPs to the lung. Interestingly, pretreatment with bivalirudin maintains lung localization. D) Additionally, bivalirudin further preserves luciferase expression of +DOTAP LNPs whereas heparin pretreatment attenuates this expression capacity. Statistics: n = 3 and the data shown represents mean ± SEM. For (C) and (D), comparisons between groups were made using a two-way ANOVA with Tukey's post-hoc test. * = p < 0.05, *** = p < 0.001, **** = p < 0.0001.
Based on this success with bivalirudin (a direct thrombin
inhibitor), we tested the effects of a similar anticoagulant,
dabigatran (Figure S15,
Supporting Information). Furthermore, we fabricated +DOTAP LNPs with a
surface-conjugated direct thrombin inhibitor, PPACK (Figure S16, Supporting Information).[41]
This might be a more convenient clinical solution than needing to
transiently anticoagulate patients with bivalirudin. Clot formation in
the lungs was also limited in mice treated with PPACK-conjugated +DOTAP
LNPs (Figure S16d, Supporting Information).
2.6 Decreasing the Size of DOTAP LNPs Prevents Fibrinogen Binding and Clotting
We serendipitously observed that the size of +DOTAP LNPs
has a positive correlation with APTT. When we increased LNP size from
100 nm to 180 nm, APTT increased by ∼1.6-fold. However, when we
decreased the LNP size to 80 nm, APTT was restored to naive levels
indicating no significant activation of the intrinsic coagulation
pathway (Figure 6a). Similarly, 80 nm +DOTAP LNPs did not significantly activate platelets, as measured by P-selectin MFI (Figure 6b).
This led us to hypothesize that the size of +DOTAP LNPs could affect
the amount of fibrinogen molecules that can be adsorbed on the LNP
surface. We tested this hypothesis first in vitro using fluorescence NTA
to probe for fibrinogen aggregation and found that 80 nm +DOTAP LNPs
generate ≈79× fewer nanoparticle-associated fibrinogen aggregates than
100 nm +DOTAP LNPs (Figure 6c,d).
Additionally, the number of fibrinogen-nanoparticle aggregates detected
with 80 nm +DOTAP LNPs is not significantly different from that under
vehicle conditions, indicating a complete absence of detectable
fibrinogen-nanoparticle aggregation. We subsequently tested how 80 nm
+DOTAP LNPs affect fibrinogen biodistributions in vivo. As previously,
we injected mice with radiolabeled fibrinogen followed by LNPs 2 min
after for a circulation time of 30 min. While 100 nm +DOTAP LNPs almost
double fibrinogen uptake in the lung, 80 nm +DOTAP LNPs do not alter
fibrinogen biodistribution or localization ratio compared to control
(Figure 6e,f).
These data validate our hypothesis that 80 nm +DOTAP LNPs do not
detectably bind to or cause aggregation of fibrinogen. Since this
interaction is associated with thrombosis, these LNPs do not activate
platelets or the intrinsic coagulation pathway (Figure 6g).
Furthermore, histological samples from the lungs of LNP-injected mice
show that 80 nm +DOTAP LNPs do not generate visible clots like 100 nm
+DOTAP LNPs (Figure 6h).
Finally, 80 nm +DOTAP LNPs still preserve their lung tropism, as shown
by their biodistribution and mRNA expression profile (30-min and 4-h LNP
circulation time respectively) (Figure 6i,j).
These data indicate that by altering the physical properties of +DOTAP
LNPs, we can modulate the binding of fibrinogen and the subsequent
initiation of clotting while maintaining lung tropism.
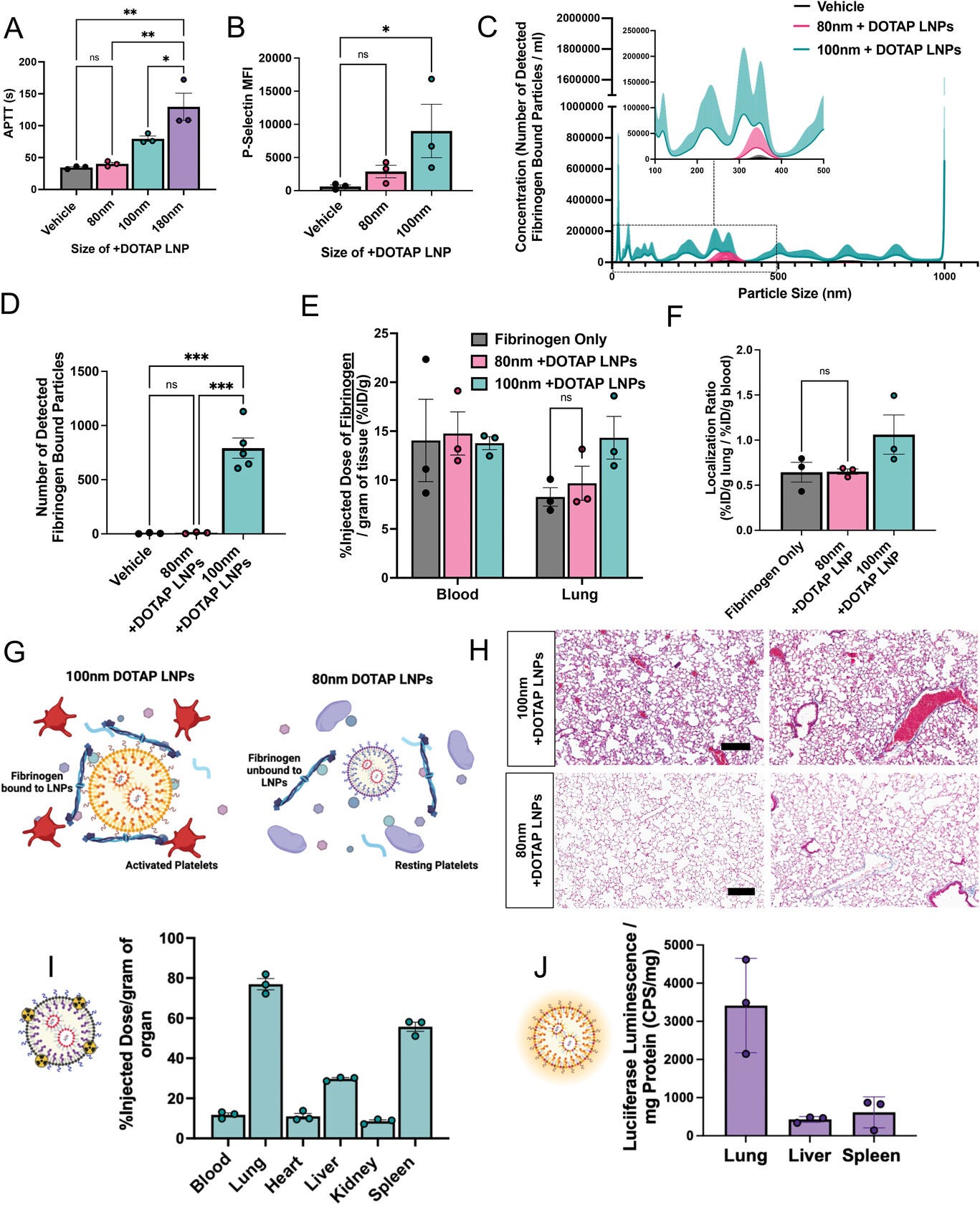
Figure 6
Open in figure viewerPowerPoint
Decreasing the size of DOTAP LNPs prevents fibrinogen binding and the resulting coagulation and platelet activation. A) +DOTAP LNP size has a positive correlation with coagulation as measured by APTT. 180 nm +DOTAP LNPs lead to a ≈1.6-fold increase in APTT compared to standard 100 nm +DOTAP LNPs while 80 nm +DOTAP LNPs maintain APTT at naive levels. B) 80 nm +DOTAP LNPs do not cause significant platelet activation as measured by P-Selectin MFI. C) Particle size versus concentration histograms from the NTA fluorescent fibrinogen binding assay comparing the number of detected fibrinogen bound particles with 80 or 100 nm +DOTAP LNPs showing that 80 nm +DOTAP LNPs bind to significantly less fibrinogen compared to 100 nm +DOTAP LNPs. Inset: Size versus concentration histogram ranging from 100–500 nm. D) Number of detected fibrinogen-bound particles from (C) showing that 80 nm +DOTAP LNPs generate ≈79× fewer fibrinogen-bound particles than 100 nm +DOTAP LNPs. The number of fibrinogen-bound particles detected with 80 nm +DOTAP LNPs is not significantly different from the vehicle. E) Amount of fibrinogen in the blood and lungs with injection of radiolabeled fibrinogen alone (vehicle) or radiolabeled fibrinogen followed by 80 or 100 nm +DOTAP LNPs. 80 nm +DOTAP LNPs do not alter fibrinogen biodistribution compared to control. F) 80 nm +DOTAP LNPs do not alter the localization ratio of fibrinogen calculated from (E) (%ID g−1 of tissue in the lung divided by that in the blood). G) Hypothesized mechanism behind the abrogation of clotting side effects below a size threshold: At a large radius of curvature (≥ 100 nm), +DOTAP LNPs present a flat enough surface to bind fibrinogen lengthwise, which then activates platelets. H) Lung histological samples reveal that 80 nm +DOTAP LNPs do not generate visible clots as is the case with 100 nm +DOTAP LNPs. I) 80 nm +DOTAP LNPs maintain their biodistribution and lung localization (10 µg mRNA per mouse, 30 min; scale bar = 200 µm) and J) express mRNA primarily in the lung measured by luciferase expression (10 µg mRNA per mouse, 4 h). Statistics: n = 3 and the data shown represents mean ± SEM. For (E), comparisons between groups were made using a two-way ANOVA with Tukey's post-hoc test. For all other graphs, comparisons between groups were made using one-way ANOVA with Tukey's post-hoc test. * = p < 0.05, *** = p < 0.001, **** = p < 0.0001.
3 Discussion
Nanoparticles are prone to attack by blood's defense
systems. Two of the major defense systems of blood, complement proteins
and immunoglobulins, have been repeatedly shown to attack nanoparticles.
But a third blood defense, clotting, is often overlooked by the field
of nanomedicine, especially the booming field of LNPs, as this is the
first report of LNP-induced thrombosis. Since intravascular clotting
leads to potentially deadly, acute thrombosis, investigation of clotting
side effects should be a standard part of IV nanomedicine development.
There have been prior studies showing thrombotic side effects of other
types of nanoparticles, but these studies have been largely neglected
with the most clinically important nanomedicines today: LNPs. As the
field of nanomedicine rapidly moves towards using charged lipids and
other physicochemically less defined mechanisms as a primary tool for
targeting, studies of clotting responses to nanomedicines, especially
LNPs, are critically timely and necessary.
Among the reasons that the LNP field had not previously
observed clotting, a major one is likely the methods used for testing
LNPs in vitro. In vitro studies of blood responses to nanoparticles
often use serum, rather than plasma. Plasma is the acellular component
of blood. Serum is prepared by depleting clotting factors from plasma,
so it is impossible to study clotting responses with serum. For
instance, fibrinogen, critical to side effects observed in our studies,
is missing from serum. Even in studies of plasma, one misses the
critical role of platelets, shown here to serve as a pro-thrombotic
surface that is activated by and amplifies the pro-coagulant effects of
cationic LNPs. Nanomedicine's focus on complement as a central source of
side effects may also miss upstream coagulation side effects: Thrombin
and plasmin activate the complement cascade. In vitro and in vivo
studies that assessed nanoparticle interactions with the acellular and
cellular components of blood involved in clotting were necessary to
evaluate the deadly side effects identified here, and similar studies
may be called for in developing other physicochemical targeting
approaches.
Here we have focused on clotting associated with LNPs that
bear a positive zeta potential, but future studies will need to
investigate other nanoparticle properties that induce clotting. In this
current study, we showed that clotting is induced by lung-tropic LNPs
containing either permanently cationic lipids (possessing quaternary
amines), or even those containing only an ionizable lipid (possessing
only tertiary amines) whose charge dynamics are such that the overall
LNP zeta potential is positive. These effects held across a wide range
of LNP lipid constituents (varying ionizable lipids, charged lipids,
etc). This strongly implicates the presence of positive zeta potential
as a nanoparticle feature that increases thrombosis risk. But we also
identified a nanoparticle feature that reduces thrombosis risk: varying
nanoparticle size had a large effect on clot induction, with a size
below ≈100 nm limiting thrombosis induced by +DOTAP LNPs in mice. Many
additional nanoparticle properties must be tested for their relationship
to clotting, including mechanical properties (Young's modulus, surface
fluidity), polymeric brush borders (e.g., PEG), and appended proteins
(e.g., antibodies used to target LNPs to specific epitopes). Such a
systematic survey, coupled with biophysics studies of nanoparticle
binding to coagulation proteins, will enable the design of safer
nanoparticles.
Just as nanoparticle properties must be studied in relation
to clotting, we must also investigate how changes in the blood itself
might affect nanoparticle-associated clotting. While these studies were
done in mice, we must now assay for nanoparticle-associated clotting in
human blood, as well as the large animals commonly used in preclinical
efficacy and toxicity studies (pig, sheep, and non-human primates),
noting that small animal models often do not capture fatal embolic side
effects that emerge in larger animals and humans. We should also
investigate how various disease states and medications that affect the
blood might predispose or protect from nanoparticle-associated clotting.
Here we showed that pre-existing inflammation worsens the magnitude of
LNP-induced lung injury, but more in-depth mechanistic studies are
required, as well as testing in other relevant disease states, such as
common procoagulant genotypes (factor V Leiden, proteins C & S
deficiency), cancers with clotting predispositions (noting that clotting
is associated with metastasis), and other inflammatory diseases.
Indeed, fibrinogen is an “acute phase reactant,” meaning that its
concentration in plasma goes up dramatically during inflammation (and
many cancers), thus making it more likely to bind to LNPs. These studies
in different disease states are essential, because many of the proposed
applications of LNPs are for cancer and inflammatory diseases, and thus
nanoparticle-induced clotting could become more prominent and
dangerous, perhaps occurring even for smaller LNPs or LNPs without
positively charged lipids.
While studying LNPs in the above thrombophilic states, it
will be essential to further optimize the clot prevention techniques
used with any LNPs that possess a positive zeta potential. Here we
showed improvement with three anti-thrombosis methods (systemic
anticoagulation, conjugating thrombin inhibitors to LNPs, and smaller
LNP diameter) in mice, but it is likely these techniques will have to be
either combined or extended to be safe in human thrombophilic states.
Thus, additional work is necessary to make positive-zeta-potential LNPs
truly safe for clinical use.
Finally, more mechanistic studies are needed to further
understand nanoparticle-induced clotting. Here we showed that fibrinogen
binds to positively charged LNPs (Figure 4b–j)
and it is likely that this is due to an electrostatic interaction
between these LNPs and the negatively charged domains of fibrinogen such
as the D and E domains, similar to other charged particles.[33, 42] We have shown that this binding leads to a large change in the conformational change of fibrinogen (Figure 4k).
We therefore hypothesize that fibrinogen binding to LNPs changes its
conformation to resemble its activated form. Such surface-bound
fibrinogen/fibrin is known to bind to GPIIB/IIIA on platelets,
immobilizing and leading to activation of the platelets. Indeed, we
showed that fibrinogen-binding +DOTAP LNPs induce platelet aggregation
and activation (Figure 3) and that depletion of fibrinogen abrogates +DOTAP LNP-induced platelet activation (Figure 4l).
But many mechanistic questions remain, such as why fibrinogen binding
is so sensitive to the LNP radius of curvature, and whether clinically
used platelet inhibitors (aspirin, clopidogrel, and especially the
GPIIB/IIIA inhibitor tirofiban) would prevent LNP-induced clotting. Such
future mechanistic studies will likely improve our techniques to
prevent LNP-associated clotting and could make physicochemical targeting
of LNPs safer and more effective.
4 Experimental Section
Materials
DOTAP, 1,2-dioleoyl-snglycero-3-phosphoethanolamine
(DOPE), cholesterol, 1,2-dimyristoyl-rac-glycero-3methoxypolyethylene
glycol-2000 (DMG-PEG 2000), dipalmitoyl phosphatidylcholine (DPPC),
1,2-dioleoyl-sn-glycero-3phosphoethanolamine-N-(TopFluor AF594) (18:1 PE
TopFluor AF 594, ammonium salt),
1,2-distearoylsn-glycero-3-phosphoethanolamine-N-diethylenetriaminepentaacetic acid
(18:0 PE-DTPA, ammonium salt),
1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS, sodium salt), and
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[azido(polyethylene
glycol)-2000] (DSPE-PEG2000-azide, ammonium salt) were purchased from
Avanti Polar Lipids.
Ionizable lipids cKK-E12, 306-N16B, SM-102,
ALC-0315, and C12-200 were purchased from Echelon Biosciences.
Anti-mouse BV421 CD41, PE-Cy7 CD62P AF700 Ly6G, and APC CD42d were
purchased from Biolegend. Indium-111 chloride (In-111) was purchased
from BWXT Medical. A modified Lowry assay kit (DC Protein Assay) was
purchased from Bio-Rad Laboratories. tPA was purchased from Millipore.
Animals
All animal studies were carried out in accordance
with the Guide for the Care and Use of Laboratory Animals (National
Institutes of Health, Bethesda, MD), and all animal protocols were
approved by the University of Pennsylvania Institutional Animal Care and
Use Committee. All animal experiments were carried out using male, 6–8
week old C57BL/6 mice (23–25 g) (The Jackson Laboratory, Bar Harbor,
ME). The mice were maintained at 22–26 °C and adhered to a 12/12 h
dark/light cycle with food and water ad libitum. For all experiments,
mice were anesthetized in a chamber with 4% isofluorane in 100% oxygen.
Nanoparticle Formulation
LNPs were formulated using the microfluidic mixing
method. An organic phase containing a mixture of lipids dissolved in
ethanol at a designated molar ratio (Table S1,
Supporting Information) was mixed with an aqueous phase (50 mM citrate
buffer, pH 4) containing luciferase mRNA that was either purchased by
TriLink (most experiments) or made in-house via in vitro transcription
(IVT)[43]
at a flow rate ratio of 1:3 and at a total lipid/mRNA weight ratio of
40:1 in a microfluidic mixing device (NanoAssemblr Ignite, Precision
Nanosystems). LNPs were dialyzed against 1 × PBS in a 10 kDa molecular
weight cut-off cassette for 2 h, sterilized through a 0.22 µm filter,
and stored at 4 °C.
To manufacture LNPs of 180 nm, the flow rate of the NanoAssemblr Ignite varied, from 6 mL min−1 (for 100 nm) to 1 mL min−1
(180 nm). The authors were unable to produce uniform LNPs < 100 nm
with TriLink mRNA, but with in-house IVT mRNA, LNPs of 80 nm were made
at a flow rate of 6 ml min−1.
Liposomes were synthesized using the thin film
hydration method. Lipids were dissolved in chloroform and combined in a
borosilicate glass tube. Chloroform and ethanol were evaporated by
blowing nitrogen over the solution until visibly dry (≈15 min) and then
placed in the tube under vacuum for > 1 h. Dried lipid films were
hydrated with 1 × PBS, pH 7.4 to a total lipid concentration of 20 mM.
The rehydrated lipid solution was vortexed and sonicated in a bath
sonicator until visually homogeneous (≈1 min each of vortexing and
sonication). The solution was then extruded 21 times through a 0.2 µm
polycarbonate filter.
Nanoparticle Characterization
Dynamic light scattering measurements of
hydrodynamic nanoparticle size, distribution, polydispersity index, and
zeta potential were made using a Zetasizer Pro ZS (Malvern Panalytical).
LNP RNA encapsulation efficiencies and concentrations were measured
using a Quant-iT RiboGreen RNA assay (Invitrogen).
Nebulized LPS Model
Mice were exposed to nebulized LPS in a
“whole-body” exposure chamber, with separate compartments for each mouse
(MPC-3 AERO; Braintree Scientific, Inc.; Braintree MA). To maintain
adequate hydration, mice were injected with 1 mL of sterile saline, 37
°C, intraperitoneally, immediately before exposure to LPS. LPS
(L2630-100 mg, Sigma Aldrich) was reconstituted in PBS to 10 mg mL−1 and stored at −80 °C until use. Immediately before nebulization, LPS was thawed and diluted to 5 mg mL−1
with PBS. LPS was aerosolized via a mesh nebulizer (Aerogen, Kent
Scientific) connected to the exposure chamber (NEB-MED H, Braintree
Scientific, Inc.). 5 mL of 5 mg mL−1 LPS was used to induce the injury. Nebulization was performed until all liquid was nebulized (≈20 min).
TAT and Blood Count Measurements
LNPs were retro-orbitally injected into naïve or
nebulized LPS-injured mice for a circulation time of 30 min. Blood was
collected from mice into tubes containing EDTA. Blood cells were
analyzed using an Abaxis VetScan HM5 Hematology Analyzer for complete
blood counts. Blood was then centrifuged at 1500 × g for 10 min and
plasma was collected and analyzed with a TAT ELISA kit (Abcam) according
to the manufacturer's instructions.
Lung Histology
LNPs were retro-orbitally injected into naïve mice
for a circulation time of 30 min. After exsanguination and perfusion via
the right ventricle with ≈5 mL of phosphate-buffered saline (PBS) at a
constant pressure of 25 cm H2O, whole lungs were inflated and fixed with
neutral buffered 10% formalin. Paraffin-embedded 5 µm lung sections
were stained and imaged with Masson's trichrome by the Pathology Core
Laboratory of Children's Hospital of Philadelphia.
In Vitro and In Vivo PT, APTT, and Time to Clot Assays
PT and APTT measurements were carried out with a
STart Analyzer (Stago) and Pacific Hemostasis reagents at a temperature
of 37 °C. In these studies, an oscillating stainless steel ball was
placed in the sample and after the formation of a clot, this ball was
attenuated and the time interval for clot formation was automatically
recorded. Blood was first collected from naïve mice and anticoagulated
with 3.2% sodium citrate at a ratio of 10:1, blood:citrate, vol:vol.
Blood samples were then centrifuged at 1500 × g for 10 min and the
plasma was collected. 50 µl of plasma was incubated with 0.5 µg of mRNA
in LNPs or a specified number of particles for 5 min. For the PT assay,
50 µl of LNP-treated plasma was diluted up to 100 µl with PBS and 200
uµl of PT reagent (Pacific Hemostasis) was added and the time to clot
was automatically measured. For the APTT assay, 50 µl of LNP-treated
plasma was diluted up to 100 µl of PBS and 100 µl of APTT-XL reagent
(Pacific Hemostasis) was added for an incubation time of 4 min. 100 µl
of 0.02 M calcium chloride was then added and the time to clot was
automatically measured. For in vivo measurements, blood was collected
from treated mice and then centrifuged at 1500 × g for 10 min to isolate
the plasma. Plasma was then analyzed as described.
For time-to-clot assays, 50 µl of plasma was placed
in the STart Analyzer with an oscillating stainless steel ball. After
recalcification with 100 µl of 0.02 M calcium chloride, the time to clot
was immediately measured based on the attenuation of the ball.
NTA Assay for Fibrinogen Aggregation with LNPs
To prepare fluorophore-labeled fibrinogen, mouse
fibrinogen (Innovative Research) was incubated with NHS ester Alexa
Fluor 488 (ThermoFisher) at 1:10 mol:mol ratio in PBS at 4 °C for 16 h.
Afterward, excess fluorophore was removed from fibrinogen by a 3-fold
passage against a 10 kDa molecular weight cut-off centrifugal filter
(Amicon) with PBS washing between passages. After fibrinogen recovery
from the centrifugal filter, spectrophotometer measurement of optical
density at 280 nm (Nanodrop) determined fluorescent fibrinogen
concentration and optical density measurement at 488 nm determined the
number of fluorophores per fibrinogen.
Immediately before experiments, LNP concentrations
were determined by NTA (Nanosight, Malvern). In a total reaction volume
of 40 µL, 4E10 LNPs were combined with 20 µL of heparinized mouse serum
(a pooled sample obtained from n = 3 mice) and fluorescent fibrinogen was doped into the solution at a final physiologically relevant concentration of 3 mg mL−1.
Fluorescent fibrinogen, serum, and LNPs were incubated in the dark at
room temperature for 10 min. Fluorescent fibrinogen was also added to
serum solutions at identical concentrations, without LNPs, verifying
that fluorescent fibrinogen did not detectably adhere to endogenous
serum components. The fibrinogen-serum-LNP reactions were terminated by
1:250 dilution in PBS and the diluted suspensions were used for
nanoparticle tracking analysis. NTA was conducted with a 488 nm
excitation laser and a 500 nm long pass filter to image and track the
Alexa Fluor 488 signal from fluorescent fibrinogen on LNPs. Automated
analysis of fluorescence nanoparticle tracking data in Malvern Nanosight
software used a uniform detection threshold of 5 for all samples. For
both fluorescence data and scattering data, three to five technical
replicates were obtained for each sample and an average of those
replicates was taken as representative of the size-concentration profile
for each sample.
Circular Dichroism
Measurements were recorded using a 1500 circular
dichroism spectrometer (Jasco) with a 1 mm path length quartz cell over a
wavelength range of 190–260 nm. Data were collected at a temperature of
37 °C with a bandwidth of 1 nm at 50 nm min−1 and a CD scale of 200mdeg/0.1dOD. Fibrinogen was diluted in PBS at a concentration of 1 mg ml−1 and for readings with LNPs, 4E11 particles were incubated with fibrinogen for 5 min before reading.
Radiolabeling and Biodistributions
For biodistribution studies, nanoparticles were traced with In-111 as previously described.[44]
Nanoparticles were produced as described above with 0.1 mol% of 18:0
PE-DTPA (a chelator containing lipid) using metal-free buffers. Trace
metals were removed from the buffers using a Chelex 100 resin, per the
manufacturer's instructions, to prevent unwanted occupancy of the
chelator. In-111 chloride was added to the nanocarriers at a specific
activity of 1 µCi of In-111 per 1 µmol of lipid. The mixture was
incubated at room temperature for 30 min. Then, unincorporated In-111
was removed using a Zeba Spin desalting column. The removal of
unincorporated In-111 was verified using thin film chromatography (TLC).
A 1 µL sample of nanoparticles was applied to the stationary phase
(silica gel strip). The strip was placed in the mobile phase of 10 mM
EDTA until the solvent front was 1 cm from the end of the strip (≈10
min). The strip was cut 1 cm above the initial sample location. In-111
chelated to the nanoparticles stayed at the origin, while unchelated
In-111 traveled with the solvent front. The activity in each section was
measured using a gamma counter. The percent of In-111 chelated to the
nanoparticles was calculated as the activity in the origin strip divided
by the total activity in both strips. For all experiments, > 95% of
In-111 was chelated to the nanoparticles.
For biodistributions of fibrinogen, the protein was
radiolabeled with iodine-125 using the Iodogen method. Tubes coated
with 100 µg of Iodogen reagent were incubated with fibrinogen (2 mg mL−1)
and Na125I (115µCi per µg protein) for 5 min on ice. Unincorporated
I-125 was removed with a Zeba column followed by TLC analysis as
described above.
For nanoparticle biodistributions, In-111 labeled
nanoparticles were injected into mice for a circulation time of 30 min.
For fibrinogen biodistributions, I-125 fibrinogen was allowed to
circulate for 2 min before the injection of nanoparticles for 30 min
before harvest. The radioactivity in each organ was then read with a
gamma counter (Wizard2, PerkinElmer).
Confocal Microscopy
Healthy mice were injected with 150 µg of
fibrinogen fluorescently labeled with AF647-NHS-Ester. Two minutes
later, mice were injected with fluorescent + or −DOTAP LNPs formulated
with 0.3 mol % of 18:1 PE TopFluor AF 594. After a 30-min circulation
the animals were sacrificed and perfused with 5 ml of cold PBS and lungs
were harvested and freshly frozen. 10 µm lung slices were cryosectioned
for imaging using a Leica TCS SP8 confocal microscope.
Luciferase Delivery
Luciferase mRNA LNPs fabricated as described above
were injected into mice for a circulation time of 4 h. Select organs
were then flash-frozen until the day of analysis or homogenized
immediately. Samples were suspended in 900 µL of homogenization buffer
(5 mM EDTA, 10 mM EDTA, 1:100 diluted stock protease inhibitor [Sigma],
and 1 × PBS, samples were then loaded with a steel bead [Qiagen], then
placed in a tissue homogenizer [Powerlyzer 24, Qiagen] using the
following settings: Speed (S) 2000 rpm, 2 cycles (C), time (T) 45 s, and
pause for 30 s). After this, 100 µL of lysis buffer (10% Triton-X 100
and PBS) was added into each tube and then allowed to incubate for 1 h
at 4 °C. After this, they were immediately transferred into fresh tubes,
and sonicated, using a point sonicator to remove in excess DNA, using
an amplitude of 30%, five cycles of 3 s on/off. After this, samples were
then centrifuged at 16 000 × g for 10 min. The resultant lysate was
either frozen or prepared for luminometry analysis. For luciferase
expression, 20 µL of undiluted sample was loaded onto a black 96
well-plate and then 100 uL luciferin solution (Promega) was added
immediately before reading on a luminometer (Wallac). Last, a Lowry
assay (Bio-Rad) was performed according to manufacturer's specifications
using diluted samples, specifically a 1:40 dilution for lung and spleen
tissues and a 1:80 dilution for liver tissues. Final luminescence
readings were then normalized based on the total protein concentration
obtained from the Lowry assay
Platelet Flow Cytometry
Platelet-rich plasma (PRP) was collected by first adding 15 µL of 10 mg mL−1
bivalirudin as well as flushing 23G collection needles with the same
solution bivalirudin, collecting between 500–1000 uL of blood per mouse.
After the blood draw, samples were then centrifuged at room temperature
(RT) at 200 × g for 10 min. After centrifugation, PRP was collected,
taking care not to disturb the resultant pellet. After collection,
samples were immediately read on a hematology analyzer (Vetscan HM2,
Abaxis) to obtain platelet count. Resultant platelets were then
aliquoted to achieve a total of 400 000 platelets per analysis with
Tyrode's buffer. These platelet samples were then incubated with 0.5µg
mRNA dose of AlexFluor 594 labeled −DOTAP or +DOTAP LNPs for 30 min at
37 °C. After incubation with nanoparticles, samples were stained with
flow cytometry antibodies against CD41, CD42d, and P-selectin and
allowed to incubate at RT for 10 min. Samples were then immediately
diluted with 350 µL of Tyrode's buffer and read on LSR Fortessa (BD
Bioscience). Analysis was then completed using FloJo and the gating
strategy employed is found in Figure S8,
Supporting Information. For fibrinogen-depleted plasma, 0.278 mg of tPA
was added per mL of platelet-free plasma, followed by 3 h incubation at
37 °C.
Statistics
All results were expressed as mean ± SEM unless
specified otherwise. Statistical analyses were performed using GraphPad
Prism 8 (GraphPad Software) * denotes p < 0.05, ** denotes p < 0.01, *** denotes p < 0.001, **** denotes p < 0.0001.
Acknowledgements
S.O.-L. and M.E.Z. contributed equally to this work.
Research reported in this publication was supported by the American
Heart Association under Grant 23PRE1014444 (to S.O.-L.), Ruth L.
Kirschstein National Research Service Award (NRSA) F31HL154662 (to
M.E.Z.) American Heart Association under Grant 916172 (to J.N.), PhRMA
Foundation under grant 2023 PFDL 1008128 (to Z.W.), and Grant NIH R01
HL157189 (to V.M., J.W.M., and J.S.B.). The data for this manuscript
were generated in the Penn Cytomics and Cell Sorting Shared Resource
Laboratory at the University of Pennsylvania and is partially supported
by the Abramson Cancer Center NCI Grant (P30 016520). The research
identifier number is RRid:SCR_022376.
Conflict of Interest
The authors declare no conflict of interest.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supporting Information
References
Citing Literature
Number of times cited according to CrossRef:
77
Xiaomin Fu, Zhongyuan Cai, Shengxiang Fu, Yingzi Cao, Haojie Gu, Rongrong Jin, Chunchao Xia, Su Lui, Bin Song, Qiyong Gong, Yingkun Guo, Hua Ai, Boronic acid group modified Mn-porphyrin nanoparticles evade macrophage uptake for lymph node metastasis diagnosis via MRI, Biomaterials, 10.1016/j.biomaterials.2026.124023, 330, (124023), (2026).
Qiu Zeng, Yuan Guo, Anyu Yang, Jun Li, Nanomedicine for venous thrombosis: Insights from an inflammatory perspective, Biomaterials, 10.1016/j.biomaterials.2025.123910, 329, (123910), (2026).
Kelsey L. Swingle, Michael J. Mitchell, Preparation of placenta-tropic mRNA lipid nanoparticles for pregnancy disorders, Nature Protocols, 10.1038/s41596-025-01325-7, (2026).
Ji Liu, Yangyang Zhang, Rui Yao, Tianyu Ma, Zhenwen Zhao, Ming Wang,
Mass Spectrometry-Assisted High-Throughput Discovery of Lung-Specific Lipid Nanoparticles for
In Vivo
Protein Delivery
, Nano Letters, 10.1021/acs.nanolett.6c00040, (2026).
Moritz Marschhofer, Siyu Chen, Müge Molbay, Benjamin Winkeljann, Ersilia Villano, Corinne Giancaspro, Alexandra Kourou, Otto Berninghausen, Susanne Rieder, Charlotte Ungewickell, Roland Beckmann, Bastian Popper, Ana Maria Torres, Anxo Vidal, Olivia M. Merkel, Simone P. Carneiro, Optimized lipid nanoparticles for pulmonary delivery of CRISPR/Cas9 targeting KRAS G12S in lung cancer, Journal of Controlled Release, 10.1016/j.jconrel.2026.114607, 391, (114607), (2026).
Erick D. Guerrero, Amogh Vaidya, Julien Santelli, Zeru Tian, Gabriela A. Pazzi, Daniel J. Siegwart, Investigating the functional contributions of phospholipids in selective organ targeting lipid nanoparticles, Biomaterials, 10.1016/j.biomaterials.2025.123671, 326, (123671), (2026).
Qin Wang, Shanshan Chen, Gang Li, Tailin Hou, Tongyue Yao, Mo Zhou, Zhuang Liu, Wei Jiang, Yucai Wang, Engineering Polymer–Lipid Integrated Nanoparticles with Quantitative Design Principles for Organ-Selective mRNA Delivery, Journal of the American Chemical Society, 10.1021/jacs.5c22504, (2026).
Yan Zong, Haiping Zhong, Yuqing Wang, Yi Lin, Linxi Zhao, Xiaoyu Wang, Jianan Cai, Haoyang Lin, Yixi Xiao, Tuo Wei, Shutao Guo, Qiang Cheng, Safe and Highly Efficient Lipid-Pro-Dexamethasone Nanoparticles for mRNA Delivery and Base Editing, Journal of the American Chemical Society, 10.1021/jacs.6c01708, 148, 9, (10225-10240), (2026).
Bookun Kim, Dajeong Kim, Choong-Min Ryu, Nanotech meets antibiotics: nucleotide antibiotics delivered by lipid nanoparticles, Frontiers in Cellular and Infection Microbiology, 10.3389/fcimb.2025.1737088, 15, (2026).
Jingjiao Li, Yu Liu, Xinghuan Ma, Sujia Liu, Chen Yang, Yuzhou Zhang, Xingdi Cheng, Yixing Wen, Shiwei Mi, Haowei Zu, Wei Li, Yuanyuan Zhao, Qing Li, Shuai Liu, Haonan Huo, Guizhi Shi, Jiaqi Lin, Xueguang Lu, Parenteral vaccination with an adjuvanted mRNA vaccine induces protective mucosal immunity against rotavirus in neonatal mice, Science Translational Medicine, 10.1126/scitranslmed.adw6105, 18, 836, (2026).
Mariah L. Arral, Kathryn A. Whitehead, Design principles of lipid nanoparticles for RNA delivery, Nature Reviews Bioengineering, 10.1038/s44222-026-00401-1, (2026).
Hao Shen, Dongwan Cheng, Liqing Liu, Min Li, Yuanyuan Zhou, Dan Bai, Peiwei Huangyang, Han Feng, RNAi therapy targeting coronavirus genomes, pulmonary delivery strategies and design principles: A review, International Journal of Biological Macromolecules, 10.1016/j.ijbiomac.2026.150223, 341, (150223), (2026).
Nicolas Marzolini, Taylor V. Brysgel, Ryan J. Rahman, Eno‐Obong Essien, Saw Y. Nwe, Jichuan Wu, Aparajeeta Majumder, Manthan N. Patel, Sachchidanand Tiwari, Carolann L. Espy, Fengyi Dong, Sabrina A. Santos‐De León, Anit Shah, Vladimir V. Shuvaev, Elizabeth D. Hood, Liam S. Chase, Drew Weissman, Jeremy B. Katzen, David B. Frank, Mariko L. Bennett, Oscar A. Marcos‐Contreras, Jacob W. Myerson, Vladimir R. Muzykantov, Sahily Reyes‐Esteves, Jacob S. Brenner, Targeting DNA‐LNPs to Endothelial Cells Improves Expression Magnitude, Duration, and Specificity, Advanced Science, 10.1002/advs.202515480, 13, 14, (2026).
Lei Yue, Xiulei Gao, Wei Qi, Lvhong Zhang, Yuefei Wang, Optimizing the targeting of lipid nanoparticles for gene therapy, Nanoscale Horizons, 10.1039/D5NH00351B, 11, 2, (334-356), (2026).
Taylor V. Brysgel, Jing Liu, Elizabeth D. Hood, Jacob S. Brenner, Vladimir R. Muzykantov, Equip
your vehicle for the right terrain: Using features of local pathology
to guide engineering of targeted drug delivery systems, Journal of Controlled Release, 10.1016/j.jconrel.2025.114412, 389, (114412), (2026).
Katrin F. Wiebe, Stefan C. Schneid, Werner Hoheisel, Wolfgang Frieß, Olivia M. Merkel, Impact
of formulation and process parameters on the stability and bioactivity
of RNA-loaded lipid nanoparticles during nebulization, European Journal of Pharmaceutical Sciences, 10.1016/j.ejps.2025.107383, 216, (107383), (2026).
Yan Zong, Yuqing Wang, Yi Lin, Qian Li, Mengyao Li, Hongyu Ren, Xiaoxia Wang, Qiang Ma, Xueliang Yu, Rongkuan Hu, Tuo Wei, Qiang Cheng, Synergistic targeted (SynTar) lipid nanoparticles enhance the efficacy and specificity of mRNA delivery in lungs, Acta Pharmaceutica Sinica B, 10.1016/j.apsb.2026.01.002, (2026).
N. D. Prasad Atmuri, Fariba Saadati, Jayesh Kulkarni, Dominik Witzigmann, Pieter R. Cullis, Marco A. Ciufolini, Design of cationic ionizable lipids for the delivery of therapeutic nucleic acids, Molecular Therapy Methods & Clinical Development, 10.1016/j.omtm.2025.101585, 33, 4, (101585), (2025).
Kara Gentry, Liming Lian, Hyejin Kim, Ozgenur Celik, Camille Jones, Ananda R. Podilapu, Avraham Shakked, David Loughrey, Ryan Zenhausern, Bora Jang, Jessie Doan, Sebastian Rudden, James E. Dahlman, Glycolipid nanoparticles target the spleen and detarget the liver without charge, Proceedings of the National Academy of Sciences, 10.1073/pnas.2409569122, 122, 45, (2025).
Edward B. Guzman, Piotr S. Kowalski, Robert S. Langer, Daniel G. Anderson, Activation of the coagulation cascade as a mechanism for selective nanoparticle-mediated RNA delivery to the endothelium in vivo, Science Advances, 10.1126/sciadv.ady2738, 11, 43, (2025).
Mahmoud M. AbdElwakil, Jeffrey Ni, Summer Ramsay-Burrough, Paul Joshua Hurst, Rebecca L. McClellan, Samuel R. Khasnavis, Yuan Jia, Sehar R. Masud, Timothy R. Blake, Adrienne Sallets, Ole A. W. Haabeth, Idit Sagiv-Barfi, Debra K. Czerwinski, Marco Herrera-Barrera, Ronald Levy, Paul A. Wender, Maya E. Kumar, Robert M. Waymouth,
In Vivo
mRNA Delivery to the Lung Vascular Endothelium by Dicationic Charge-Altering Releasable Transporters
, Journal of the American Chemical Society, 10.1021/jacs.5c06654, 147, 44, (40146-40157), (2025).
Qi Wang, Sikun Xiong, Ranbo Zhao, Junxi Chen, Sixuan Li, Wei Jiang, Ling Zhang, Organ‐Targeted and Organelle‐Targeted Liposome Gene Vector Construction, Advanced Healthcare Materials, 10.1002/adhm.202502254, 15, 4, (2025).
Hanqin Zhao, Yuxi Gao, Yibo Qi, An Ziyue, Minhui Li, Jie Chen, Sheng Ma, Wantong Song, Xuesi Chen, Mass-Ratio-Controlled Organ-Selective Phosphatidyl Polymer Carrier for In Vivo Targeted mRNA Delivery, ACS Nano, 10.1021/acsnano.5c12386, 19, 40, (35797-35811), (2025).
Yuqin Men, David O. Popoola, Zhi Cao, Yiran Li, Stephan Wilkens, Yong Teng, Qinghe Meng, Marc Hershenson, Yamin Li, Sulfonium lipid nanoparticles for intranasal mRNA delivery to lung epithelial and immune cells, Acta Biomaterialia, 10.1016/j.actbio.2025.08.032, 205, (616-633), (2025).
Hyun-Jin Kim, Ngoc Duy Le, Hyun-Ji Oh, Beomsu Kim, Eunjae Yoo, Jeonghwan Kim, Hyungshin Yim, Lipid nanoparticle-assisted mRNA therapy for cancer treatment, Applied Physics Reviews, 10.1063/5.0247029, 12, 3, (2025).
Andrew R. Hanna, David A. Issadore, Michael J. Mitchell, High-throughput platforms for machine learning-guided lipid nanoparticle design, Nature Reviews Materials, 10.1038/s41578-025-00831-0, 11, 1, (50-64), (2025).
Tie Chang, Yifan Zheng, Mingrui Jiang, Siqi Jia, Jianbo Bai, Zhi Zheng, Shixin Li, Jia Guo, Yue Wang, Yiting Wang, Haixia Liu, Jianlin Liu, Liangyu Ni, Xingdi Cheng, Shuai Liu, Huijuan Zhang, Wei Pi, Feng Lin, Shiyi Liu, Weijian Wang, Guannan Wang, Leyun Wang, Lei Miao, Xueguang Lu, Ziqing Deng, Bing Bai, Zhao Qin, Huajian Gao, Yue Shao, Peptide codes for organ-selective mRNA delivery, Nature Materials, 10.1038/s41563-025-02331-6, 25, 1, (146-159), (2025).
Rose Razavi, Michael Kegel, Jenna Muscat-Rivera, Drew Weissman, Jilian R. Melamed, Harnessing mRNA-lipid nanoparticles as innovative therapies for autoimmune diseases, Molecular Therapy Methods & Clinical Development, 10.1016/j.omtm.2025.101566, 33, 3, (101566), (2025).
Cedric A. Brimacombe, Jayesh A. Kulkarni, Miffy H.Y. Cheng, Kevin An, Dominik Witzigmann, Pieter R. Cullis, Rational design of lipid nanoparticles for enabling gene therapies, Molecular Therapy Methods & Clinical Development, 10.1016/j.omtm.2025.101518, 33, 3, (101518), (2025).
Aljoscha Gabelmann, Achim Biesel, Brigitta Loretz, Claus-Michael Lehr, Exploring the future of mRNA delivery: Beyond lipid nanoparticles, Biochemical and Biophysical Research Communications, 10.1016/j.bbrc.2025.152347, 778, (152347), (2025).
Moritz Marschhofer, Simone P. Carneiro, Olivia M. Merkel, Recent advances in the nanoplatform-mediated delivery of gene editing technologies to the lungs, Nanomedicine, 10.1080/17435889.2025.2550231, 20, 22, (2713-2715), (2025).
Zhijian Li, Aloysius Ee, Laura Amaya, Jennifer L. Hamad, Pavan K. Yadav, Sean K. Wang, Howard Y. Chang, Paul A. Wender, Discrete Immolative Guanidinium Transporters deliver mRNA to specific organs and red blood cells, Nature Communications, 10.1038/s41467-025-62200-3, 16, 1, (2025).
Qiancheng Gu, Huaqian Xue, Zhiyun Liu, Jiameng Rao, Lingyao Zeng, Chen Zhang, Lanjie Lei, Liyun Shi, Developing Biomaterial‐Based mRNA Delivery System for Lung Disease Treatment, Advanced Science, 10.1002/advs.202505413, 12, 34, (2025).
Bianka Golba, Irene De Franceschi, Zifu Zhong, Martijn J. Schuijs, Claudia M. Brenis Gomez, Heleen Lauwers, Benoit Louage, Iasona Sheshi, Nezha Badi, Bruno G. De Geest, Filip E. Du Prez, Combinatorial Synthesis and Evaluation of Trialkyl Galloyl Amidoamine Ionizable Lipids for mRNA Formulation, Journal of the American Chemical Society, 10.1021/jacs.5c09030, 147, 30, (26934-26943), (2025).
Victor Marx, Yuki Mochida, Shigehito Osawa, Theofilus A. Tockary, Jumpei Norimatsu, Hidetomo Yokoo, Makoto Oba, Satoshi Uchida, Poly(sarcosine) and Poly(2-ethyl-2-oxazoline) Tethered To mRNA Provide Stealth Properties To mRNA Polyplexes, ACS Applied Nano Materials, 10.1021/acsanm.5c00636, 8, 27, (13628-13638), (2025).
Yunhao You, Shaoqiu Leng, Jie Shi, Han Yang, Mingzheng Chang, Qingliang Ma, Dapeng Zhang, Haochen Sun, Lianlei Wang, Zhiliang Gao, Jiwei Cui, Xinyu Liu, Integration of Bone-Targeted Delivery and Crosstalk Modulation of Liver-Bone Axis for Improved Osteoporosis Therapy, ACS Nano, 10.1021/acsnano.5c05460, 19, 26, (23955-23968), (2025).
Roy van der Meel, Paul A. Wender, Olivia M. Merkel, Irene Lostalé-Seijo, Javier Montenegro, Ali Miserez, Quentin Laurent, Hanadi Sleiman, Paola Luciani, Next-generation materials for nucleic acid delivery, Nature Reviews Materials, 10.1038/s41578-025-00814-1, 10, 7, (490-499), (2025).
Zeying Cao, Shilin Zhou, Yanli Zhao, Qian Wu, Chenxi Huang, Qin Nie, Ting Xiong, Xiaoxu Hao, Shuo Zhang, Haojie Bao, Caifen Wang, Zongneng Xie, Jiwen Zhang, Xianzhen Yin, Tracheal
Targeted Nanogrid Delivery Systems of Dexamethasone Visualized by
Single-Particle Tracing and Multiscale Pathological Mapping, ACS Nano, 10.1021/acsnano.5c06694, 19, 25, (23414-23430), (2025).
Sijia Xu, Zhenzhen Hu, Fenglin Song, Ying Xu, Xuexiang Han, Lipid nanoparticles: Composition, formulation, and application, Molecular Therapy Methods & Clinical Development, 10.1016/j.omtm.2025.101463, 33, 2, (101463), (2025).
Yuichi Suzuki, Mai Yakuwa, Mina Sato, Eleni Samaridou, Moritz Beck-Broichsitter, Masatoshi Maeki, Manabu Tokeshi, Yuma Yamada, Hideyoshi Harashima, Yusuke Sato, Splenic B cell-targeting lipid nanoparticles for safe and effective mRNA vaccine delivery, Journal of Controlled Release, 10.1016/j.jconrel.2025.113687, 382, (113687), (2025).
Hannah Lukeman, Elizabeth Chan, James Triccas, Targeting tuberculosis with LNP-mRNA vaccines: opportunities, challenges and future directions, Microbiology Australia, 10.1071/MA25017, 46, 2, (48-51), (2025).
Paul J. L. Schürmann, Stijn P. E. Breda Vriesman, Jose A. Castro‐Alpízar, Sander A. A. Kooijmans, Edward E. S. Nieuwenhuis, Raymond M. Schiffelers, Sabine A. Fuchs, Therapeutic Application of mRNA for Genetic Diseases, WIREs Nanomedicine and Nanobiotechnology, 10.1002/wnan.70019, 17, 3, (2025).
Serena Omo-Lamai, Marco E. Zamora, Bernhard K. Mueller, Jessica C. Casciano, Jacob S. Brenner, RNA–lipid nanoparticles for acute critical illnesses, Nature Reviews Bioengineering, 10.1038/s44222-025-00314-5, 3, 9, (775-790), (2025).
Jumpei Norimatsu, Rashida Hashim, Theofilus A. Tockary, Muhammad Shahid Nazir, Yuki Mochida, Satoshi Uchida, Polycations Improve Intracellular Stability of mRNA Delivered Using Lipid Nanoparticles for Prolonged Protein Expression, ACS Applied Nano Materials, 10.1021/acsanm.5c00633, 8, 21, (10898-10906), (2025).
Aida López Espinar, Lianne M. Mulder, Mohamed Elkhashab, Zahra Khan, Mariusz Czarnocki-Cieciura, Maria R. Aburto, Sonja Vucen, Piotr S. Kowalski, Tailoring Alkyl Side Chains of Ionizable Amino-Polyesters for Enhanced In Vivo mRNA Delivery, ACS Applied Bio Materials, 10.1021/acsabm.5c00116, 8, 5, (3958-3971), (2025).
Shuang Liang, Shuying Gao, Shunli Fu, Shijun Yuan, Jinhu Liu, Man Liang, Leiqiang Han, Zipeng Zhang, Yongjun Liu, Na Zhang, Screening
Natural Cholesterol Analogs to Assemble Self‐Adjuvant Lipid
Nanoparticles for Antigens Tagging Guided Therapeutic Tumor Vaccine, Advanced Materials, 10.1002/adma.202419182, 37, 27, (2025).
Pengyu Zhu, Yongsheng Li, Dapeng Zhang, One‐Component Ionizable Amphiphilic Janus Dendrimers for Targeted mRNA Delivery, Angewandte Chemie International Edition, 10.1002/anie.202505304, 64, 22, (2025).
Pengyu Zhu, Yongsheng Li, Dapeng Zhang, One‐Component Ionizable Amphiphilic Janus Dendrimers for Targeted mRNA Delivery, Angewandte Chemie, 10.1002/ange.202505304, 137, 22, (2025).
Haiyang Yong, Zhili Li, Lixin Lin, Shuai Liu, Chao Feng, Songmei Geng, Tao Fu, Zhengju Chen, Dezhong Zhou, Aminolysis of Highly Branched Poly(β-amino ester)s for Efficient mRNA Delivery, Chemistry of Materials, 10.1021/acs.chemmater.4c03450, 37, 8, (2827-2835), (2025).
Brittany J. Heiser, Arian Veyssi, Debadyuti Ghosh, Recent strategies for enhanced delivery of mRNA to the lungs, Nanomedicine, 10.1080/17435889.2025.2485669, 20, 9, (1043-1069), (2025).
Robby Zwolsman, Youssef B. Darwish, Ewelina Kluza, Roy Meel, Engineering Lipid Nanoparticles for mRNA Immunotherapy, WIREs Nanomedicine and Nanobiotechnology, 10.1002/wnan.70007, 17, 2, (2025).
Rebecca M. Haley, Marshall S. Padilla, Rakan D. El-Mayta, Ryann A. Joseph, Jesse A. Weber, Christian G. Figueroa-Espada, Alvin J. Mukalel, Adele S. Ricciardi, Rohan Palanki, Hannah C. Geisler, Matthew T. Jester, Beverly L. Davidson, Michael J. Mitchell,
Lipid Nanoparticles for
In Vivo
Lung Delivery of CRISPR-Cas9 Ribonucleoproteins Allow Gene Editing of Clinical Targets
, ACS Nano, 10.1021/acsnano.4c16617, 19, 14, (13790-13804), (2025).
Zhefan Yuan, Sijin Luozhong, Ruoxin Li, Wenchao Gu, Yu Chen, Dani Bhashyam, Rachel Lai, Shaoyi Jiang, Transient Macrophage Depletion Circumvents Scavenging and Redirects Biodistribution of mRNA-Lipid Nanoparticles, ACS Nano, 10.1021/acsnano.5c02001, 19, 14, (14422-14433), (2025).
Hannah C. Safford, Cecilia F. Shuler, Hannah C. Geisler, Ajay S. Thatte, Kelsey L. Swingle, Emily L. Han, Amanda M. Murray, Alex G. Hamilton, Hannah M. Yamagata, Michael J. Mitchell, Probing the Role of Lipid Nanoparticle Elasticity on mRNA Delivery to the Placenta, Nano Letters, 10.1021/acs.nanolett.4c06241, 25, 12, (4800-4808), (2025).
Yingying Shi, Jiapeng Mao, Sijie Wang, Siyao Ma, Lihua Luo, Jian You, Pharmaceutical strategies for optimized mRNA expression, Biomaterials, 10.1016/j.biomaterials.2024.122853, 314, (122853), (2025).
Xiaoyan He, Runyuan Wang, Yan Cao, Yan Ding, Yan Chang, Haoru Dong, Rong Xie, Guisheng Zhong, Huiying Yang, Jianfeng Li, Lung‐Specific mRNA Delivery by Ionizable Lipids with Defined Structure‐Function Relationship and Unique Protein Corona Feature, Advanced Science, 10.1002/advs.202416525, 12, 14, (2025).
Manthan N. Patel, Sachchidanand Tiwari, Yufei Wang, Sarah O’Neill, Jichuan Wu, Serena Omo-Lamai, Carolann Espy, Liam S. Chase, Aparajeeta Majumder, Evan Hoffman, Anit Shah, András Sárközy, Jeremy Katzen, Norbert Pardi, Jacob S. Brenner, Safer non-viral DNA delivery using lipid nanoparticles loaded with endogenous anti-inflammatory lipids, Nature Biotechnology, 10.1038/s41587-025-02556-5, 44, 1, (79-89), (2025).
Marshall S. Padilla, Kaitlin Mrksich, Yiming Wang, Rebecca M. Haley, Jacqueline J. Li, Emily L. Han, Rakan El-Mayta, Emily H. Kim, Sofia Dias, Ningqiang Gong, Sridatta V. Teerdhala, Xuexiang Han, Vivek Chowdhary, Lulu Xue, Zain Siddiqui, Hannah M. Yamagata, Dongyoon Kim, Il-Chul Yoon, James M. Wilson, Ravi Radhakrishnan, Michael J. Mitchell, Branched
endosomal disruptor (BEND) lipids mediate delivery of mRNA and
CRISPR-Cas9 ribonucleoprotein complex for hepatic gene editing and T
cell engineering, Nature Communications, 10.1038/s41467-024-55137-6, 16, 1, (2025).
Chao Liu, Yuhao Jiang, Wenliang Xue, Jinyu Liu, Zihao Wang, Jianhong Luo, Xinsong Li, Ionizable Cholesterol Analogs as the Fifth Component of Lipid Nanoparticles for Selective Targeting Delivery of mRNA, ACS Applied Materials & Interfaces, 10.1021/acsami.4c14203, 17, 3, (4416-4425), (2025).
Yuduo Gao, Luwei Zhang, Ziyue Wang, Hao Bai, Chengfan Wu, Qi Shuai, Yunfeng Yan, Enhanced Pulmonary and Splenic mRNA Delivery Using DOTAP-Incorporated Poly(β-Amino Ester)-Lipid Nanoparticles, Biomacromolecules, 10.1021/acs.biomac.4c01445, 26, 1, (623-634), (2025).
Mahmoud A. Younis, Yusuke Sato, Seigo Kimura, Hideyoshi Harashima, A
new strategy for the extrahepatic delivery of lipid-based
nanomedicines: a protein corona-mediated selective targeting system
based on an ionizable cationic lipid library, RSC Pharmaceutics, 10.1039/D5PM00079C, 2, 5, (982-1002), (2025).
Lauren Healy, Breanna Y. Seto, Haissi Cui, Bowen Li, Non-viral mRNA delivery to the lungs, Biomaterials Science, 10.1039/D5BM00322A, 13, 11, (2871-2882), (2025).
Agatha M. Pelosine, Anderson F. Sepulveda, Luccas C. T. de Jesus, Juliana Marchi, Daniele R. de Araujo, Nanomaterials in hemostasis: Modulating pathways and mechanisms with bioactive materials, Advanced Nanomaterials in Biomedical Implants: Processing, Structures, Properties and, Applications, 10.1016/B978-0-443-27378-0.00013-1, (345-366), (2025).
Hao Tang, Hanbing Wang, Zhihua Gan, Zhenshan Ding, Qingsong Yu, Engineering the Hydrophilic–Hydrophobic Interface of Polymeric Micelles by Cationic Blocks for Enhanced Chemotherapy, ACS Applied Materials & Interfaces, 10.1021/acsami.4c17024, 16, 50, (69011-69027), (2024).
Zhaoming Chen, Yuexia Yang, Xinyu Qiu, Hao Zhou, Rui Wang, Hu Xiong, Crown-like Biodegradable Lipids Enable Lung-Selective mRNA Delivery and Dual-Modal Tumor Imaging In Vivo, Journal of the American Chemical Society, 10.1021/jacs.4c14500, 146, 49, (34209-34220), (2024).
Colton Strong, Jerry Leung, Emma Kang, Katherine E. Badior, Madelaine Robertson, Nicolas Pereyra, Elyn M. Rowe, Amanda Wietrzny, Brenda Ma, Zechariah Noronha, Deaglan Arnold, Marco A. Ciufolini, Dana V. Devine, Eric Jan, Pieter R. Cullis, Christian J. Kastrup, Genetic engineering of transfusable platelets with mRNA-lipid nanoparticles is compatible with blood banking practices, Blood, 10.1182/blood.2024024405, 144, 21, (2223-2236), (2024).
Didar Baimanov, Zhuda Song, Zehao Zhang, Longxing Sun, Qingxi Yuan, Wanxia Huang, Yuxi Gao, Liming Wang, Zhiyong Zhang, Preferred Lung Accumulation of Polystyrene Nanoplastics with Negative Charges, Nano Letters, 10.1021/acs.nanolett.4c03245, (2024).
Yuzheng Zhou, Yujie Liao, Lujie Fan, Xiafei Wei, Qiang Huang, Chuwei Yang, Wei Feng, Yezi Wu, Xiang Gao, Xiaotong Shen, Jian Zhou, Zanxian Xia, Zheng Zhang, Lung‐Targeted
Lipid Nanoparticle‐Delivered siUSP33 Attenuates SARS‐CoV‐2 Replication
and Virulence by Promoting Envelope Degradation, Advanced Science, 10.1002/advs.202406211, 11, 42, (2024).
Mincheol Jang, Kyunghwan Yeom, Junhee Han, Erinn Fagan, Ji-Ho Park, Inhalable mRNA Nanoparticle with Enhanced Nebulization Stability and Pulmonary Microenvironment Infiltration, ACS Nano, 10.1021/acsnano.4c05653, 18, 35, (24204-24218), (2024).
Elizabeth R. Webster, Nicole E. Peck, Juan Diego Echeverri, Shima Gholizadeh, Wei-Lun Tang, Rinette Woo, Anushtha Sharma, Weiqun Liu, Chris S. Rae, Adrienne Sallets, Gowrisudha Adusumilli, Kannan Gunasekaran, Ole A. W. Haabeth, Meredith Leong, Ronald N. Zuckermann, Samuel Deutsch, Colin J. McKinlay, Discovery
of a Peptoid-Based Nanoparticle Platform for Therapeutic mRNA Delivery
via Diverse Library Clustering and Structural Parametrization, ACS Nano, 10.1021/acsnano.4c05513, 18, 33, (22181-22193), (2024).
David O. Popoola, Zhi Cao, Yuqin Men, Xinyuan Li, Mariano Viapiano, Stephan Wilkens, Juntao Luo, Yong Teng, Qinghe Meng, Yamin Li, Lung-Specific mRNA Delivery Enabled by Sulfonium Lipid Nanoparticles, Nano Letters, 10.1021/acs.nanolett.4c01854, 24, 26, (8080-8088), (2024).
Tomoaki Kurosaki, Yuki Ueda, Yuka Kato, Mikiro Nakashima, Takashi Kitahara, Hitoshi Sasaki, Yukinobu Kodama, Effect of a novel siRNA delivery system, siRNA ternary complex, on melanoma lung metastasis, Journal of Drug Targeting, 10.1080/1061186X.2024.2362361, 32, 7, (848-854), (2024).
Yan Liang, Chenlu Xu, Jingge Zhang, Shuguang Li, Menghao Yin, Yinchao Li, Hua Gao, Kaixiang Zhang, Simultaneously
supplementing Wtp53 and degrading Mutp53 using a virus-mimicking mRNA
delivery system to restore P53's autonomous anti-cancer function, Nano Research, 10.26599/NR.2025.94907529, 18, 6, (94907529), (2024).
Sofie Meulewaeter, Ilke Aernout, Joke Deprez, Yanou Engelen, Margo De Velder, Lorenzo Franceschini, Karine Breckpot, Serge Van Calenbergh, Caroline Asselman, Katie Boucher, Francis Impens, Stefaan C. De Smedt, Rein Verbeke, Ine Lentacker, Alpha-galactosylceramide improves the potency of mRNA LNP vaccines against cancer and intracellular bacteria, Journal of Controlled Release, 10.1016/j.jconrel.2024.04.052, 370, (379-391), (2024).
Marco E. Zamora, Serena Omo-Lamai, Manthan N. Patel, Jichuan Wu, Evguenia Arguiri, Vladmir R. Muzykantov, Jacob W. Myerson, Oscar A. Marcos-Contreras, Jacob S. Brenner, Combination of Physicochemical Tropism and Affinity Moiety Targeting of Lipid Nanoparticles Enhances Organ Targeting, Nano Letters, 10.1021/acs.nanolett.3c05031, (2024).
Sarah S. Nasr, Yahya Cheema, Alexa Stern, Owen Tabah, Stephanie Poore, Gregg A. Duncan, Tissue‐specific gene delivery approaches, Bioengineering & Translational Medicine, 10.1002/btm2.70125.
Xinfang Zhang, Zhouhua Zhong, Peimeng Zhan, Tan Ye, Yang Lan, Haochun Zhan, Zhuo Xie, Muyuan Chai, Ke Zheng, Zhengmei Lin, Shuheng Huang, Xuetao Shi, Adhesive
Microneedle‐Nanosheets With Temporally Orchestrated Antimicrobial,
Immunomodulatory, and Osteogenic Functions for Periodontal Regeneration, Advanced Materials, 10.1002/adma.72919.
The Rapley prepint is important so will likely be suppressed.
MarcGirardot in his Bolus Theory series on his <covidmythbuster.substack.com> proposes that the Covid vaxx SAE's were due to intravascular injection. This would align with your cationic LNP work, and also explain why so many believers think their covid shots were helpful- the needle actually did only stab muscle tissue.